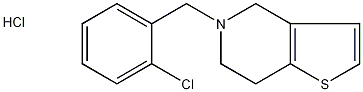
结构式
| 物竞编号 | 0TPV |
|---|---|
| 分子式 | C14H14ClNSHCl |
| 分子量 | 300.25 |
| 标签 | 盐酸噻氯匹定, 5-(o-Chlorobenzyl)-4,5,6,7-tetrahydrothieno(3,2-c)pyridine, 药物 |
编号系统
CAS号:53885-35-1
MDL号:MFCD00079606
EINECS号:258-837-4
RTECS号:XJ9089100
BRN号:暂无
PubChem号:24900396
物性数据
1. 性状:未确定
2. 密度(g/mL,25ºC):未确定
3. 相对蒸汽密度(g/mL,空气=1): 未确定
4. 熔点(ºC):205
5. 沸点(ºC):未确定
6. 沸点(ºC,5mm hg):未确定
7. 折射率:未确定
8. 闪点(°F):未确定
9. 比旋光度(ºC):未确定
10. 自燃点或引燃温度(ºC): 未确定
11. 蒸气压(kPa,20ºC):未确定
12. 饱和蒸气压(kPa,60ºC):未确定
13. 燃烧热(KJ/mol):未确定
14. 临界温度(ºC):未确定
15. 临界压力(KPa):未确定
16. 油水(辛醇/水)分配系数的对数值:未确定
17. 爆炸上限(%,V/V):未确定
18. 爆炸下限(%,V/V):未确定
19. 溶解性:溶解性:易溶于冰乙酸、乙醇或水,溶于95%乙醇、甲醇或氯仿,微溶于正丁醇, 不溶于乙醚。
毒理学数据
LD50(g/kg):小鼠(24h),静脉注射=55,经口>300
生态学数据
对水是稍微有危害的不要让未稀释或大量的产品接触地下水、水道或者污水系统,若无政府许可,勿将材料排入周围环境
分子结构数据
暂无
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:1
3.氢键受体数量:2
4.可旋转化学键数量:2
5.互变异构体数量:无
6.拓扑分子极性表面积31.5
7.重原子数量:18
8.表面电荷:0
9.复杂度:261
10.同位素原子数量:0
11.确定原子立构中心数量:0
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:2
性质与稳定性
如果遵照规格使用和储存则不会分解,未有已知危险反应
贮存方法
保持贮藏器密封、储存在阴凉、干燥的地方,确保工作间有良好的通风或排气装置
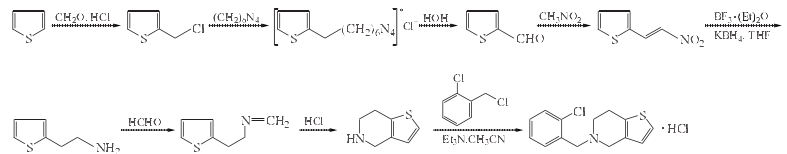
合成方法
1. 2-氯甲基噻吩的制备
在反应瓶中加入噻吩42g(39.2ml,0.5mol)和浓盐酸20ml,用冰盐浴冷却,在剧烈搅拌下使快速的HCl气体气流连续地通入混合物.当温度到达0ºC时,滴加37%甲醛水溶液50ml(0.666mol),控制加入速度,使温度保持在5ºC以下,需30~50min加完.当全部甲醛液加完后,将反应混合物用乙醚(50ml×3)提取,合并乙醚提取液,依次用水和饱和NaHCO3溶液洗涤,无水CaCl2干燥,过滤,滤液蒸除溶剂,剩余物减压蒸馏(应采用合适的分馏柱),收集73~75ºC/2.26 kPa(17mmHg)馏分,得无色油状液体2-氯甲基噻吩25.7~26.7g,收率40%~41%.
2. 氯化(2-噻吩甲基)六次甲基四铵的制备
在反应瓶中加入上步反应产物2-氯甲基噻吩67g(0.5mol)、氯仿400ml和六次甲基四胺70g(0.5mol),加热至回流,搅拌缓和回流反应30min.冷却,过滤,滤饼用冷氯仿100l洗涤,压干抽干后,晾干,得氯化(2-噻吩甲基)六次甲基四铵128~136g,收率94%~99%,为白色粉末产物.
3. 2-噻吩甲醛的制备
在2000ml圆底烧瓶中加入400ml温水和上步制
备的化合物氯化(2-噻吩甲基)六次甲基四铵一批量(128~136g),把烧瓶装配成水蒸气蒸馏装置,通入水蒸气至所有的醛被蒸出为止(蒸出所有的2-噻吩甲醛要收集大约1.5L馏出液).馏出液冷却后,加6mol/L乙酸10ml(如果2-噻吩甲醛要贮存一段时间,应加入少量对苯二酚),含醛馏出液用乙醚提取0次,每次用乙醚100ml.乙醚溶液用无水氯化钙干燥,过滤,滤液蒸至体积减少到500ml左右,然后将此溶液转移到100ml克氏烧瓶中,蒸去乙醚,再减压蒸馏,收集89~91ºC/2.799kPa(21mmHg)馏分,得无色油状物2-噻吩甲醛27~30g,收率48%~53%.
4. 2-硝基乙烯噻吩的制备
在反应瓶中加入上步制备的化合物2-噻吩甲醛56g(0.5mol)、硝基甲烷61g(1mol)、甲醇1000ml,用冰盐浴冷却至0ºC以下,搅拌滴加40%氢氧化钠水溶液100g(1mol),滴加过程中维持温度在5ºC以下,约2h滴毕,继续搅拌4h.加入水300ml ,在搅拌下慢慢倒入含浓盐酸100ml的冰水中,析出黄色沉淀.过滤,水洗,干燥,得黄色固体2-硝基乙烯噻吩54g,收率70%,mp80~82ºC.
5. 2-噻吩乙胺的制备
在反应瓶中加入硼氢化钾54g(1mol)、四氢呋喃(THF)500ml,搅拌降温至0ºC,于0ºC慢慢滴加三氟化硼乙醚液150ml(1.2mol),加毕,于0~5ºC下搅拌反应2h.再搅拌滴加化合物2-硝基乙烯噻吩31g(0.2mol)的THF300ml溶液,约1h滴毕.于室温下搅拌反应24h.加入甲苯500ml.蒸馏,当内温达到90ºC时,改成回流,搅拌回流1h.冷至0ºC.加水100ml,用1mol/L盐酸500ml慢慢酸化.加热至80ºC,搅拌1h.冷至40~50ºC.分取水层,甲苯层用1mol/L盐酸200ml洗,合并水层,用NaOH溶液调pH至13.用二氯甲烷(500ml×3)提取,合并提取液,用水200ml洗涤,无水MgSO4干燥,过滤,滤液浓缩回收二氯甲烷,剩余物为油状物2-噻吩乙胺约19.5g,收率80%.
6. 2-噻吩乙基亚甲胺的制备
在反应瓶中加入上步制备的化合物2-噻吩乙胺12.7g(0.1mol),在搅拌下于室温滴加36%~37%甲醛水溶液10g(0.12mol).滴毕,搅拌回流反应3h.冷却,#用二氯甲烷(150ml×3)提取,合并提取液,用水50ml洗涤,无水MgSO4干燥,过滤,滤液浓缩,得淡黄色油状物2-噻吩乙基亚甲胺12.8g,收率92%.
7. 4,5,6,7-四氢噻吩并[3,2-c]吡啶的制备
在反应瓶中加入上步制备的化合物2-噻吩乙基亚甲胺13.9g(0.1mol)和6mol/L盐酸28ml,于室温搅拌反应6h.加入NaOH水溶液调pH至13,用二氯甲烷(200ml×3)提取,合并有机层,用水100ml洗涤,用无水MgSO4干燥.过滤,滤液浓缩回收二氯甲烷,得浅黄色油状物4,5,6,7-四氢噻吩并[3,2-c]吡啶粗品13.9g,收率100%.无需纯化,直接用于噻氯匹定的合成.
8. 2-氯氯苄的制备
在反应瓶中加入邻氯甲苯108g(0.854mol)、过氧化苯甲酰0.4g、三氯化磷0.3g,在搅拌和光照下加热至90~95ºC,通入干燥的氯气,维持反应液温度在95~100ºC,至反应液的相对密度达1.20~1.22即停止通氯气.冷却,用温水洗涤反应液数次后,用无水Na2SO4干燥,过滤,将滤液减压蒸馏,收集93~98ºC/1.33kPa馏分,得无色液体2-氯氯苄11.2g,收率82%.
9. 5-[(2-氯苯基)甲基]-4,5,6,7-四氢噻吩并[3,2-c]吡啶盐酸盐(盐酸噻氯匹定)的合成
在反应瓶中加入第七步制备的化合物4,5,6,7-四氢噻吩并[3,2-c]吡啶13.9g(0.1mol)、上步制备的化合物2-氯氯苄16.1g(0.1mol)、三乙胺15g(0.15mol)和乙腈150ml,于室温下搅拌反应4h.减压浓缩回收乙腈,加入甲苯100ml和水100ml,充分搅拌后静置分层,分取甲苯层,水层用甲苯(50ml×3)提取,合并甲苯液,用水(50ml×3)洗涤,无水MgSO4干燥.过滤,滤液用氯化氢异丙醇溶液调pH至2,搅拌30min.放冷,析出固体,过滤,得浅黄色固体.用无水乙醇重结晶,得白色固体5-[(2-氯苯基)甲基]-4,5,6,7-四氢噻吩并[3,2-c]吡啶盐酸盐(盐酸噻氯匹定)23g,收率75%,mp206~207ºC.
用途
本品为抗血栓药#可抑制ADP胶原、凝血酶、AA和PG内过氧化物等引起的血小板凝集,其抑制作用可维持整个血小板生存期.作用机制主要是抑制Tx合成酶,并活化AC增加血小板内cAMP水平,从而抑制血小板凝集.其次,本品还能降低纤维蛋白原浓度、全血黏度和血浆黏度以及增强红细胞变形性,使微循环阻力降低,血流速度和血流量增加,从而改善微循环功能.临床上用于防治缺血性脑血管障碍、慢性四肢动脉闭塞症、血栓闭塞性血管炎以及动脉粥样硬化和血栓栓塞引起的心绞痛、脑卒中等有良好的效果.对防治血管血栓栓塞性脑卒中的复发、心肌梗死等,本品优于独用阿司匹林或阿司匹林合用潘生丁的防治效果.此外,本品可代替肝素用于血液透析,以减少肝素副作用的发生.
安全信息
危险运输编码:暂无
危险品标志: 有害
有害
安全标识:S36
危险标识:R22
文献
【1】 Merck Index.13th:9506. 【2】 Leblondel G,et al.Biochem Pharmacol,1978,27:2099. 【3】 O’Brien J R,et al.Thromb Res,1978,13:245. 【4】 Bruno J J,et al. Thromb Res,1983,(Suppl 4):59. 【5】 Akashi A,et al.Arzneimittel-Forsch,1980,30:409,415. 【6】 Thebault J J,et al.J Int Med Res,1977,5:405. 【7】 Lecrubier C,et al.Therapie,1977,32:189. 【8】 Katsumura T,et al.Angiology,1982,33:357. 【9】 Saltiel E,et al.Drugs,1987,34:222-262. 【10】 DE,2404308.1974. 【11】 US,4051141.1977. 【12】 US,4127580.1978. 【13】 刘秀杰.中国医药工业杂志,1990,21:451. 【14】 EP,342118.1989. 【15】 日本公开特许86-221184. 【16】 日本公开特许87-103088. 【17】 DE,2604248.1976. 【18】 Dressler M L,et al.J Heterocycle Chem,1970,7:1257. 【19】 罗明生主编.现代临床药物大典.成都:四川科学技术出版社,2001:735. 【20】 四川美康医药软件研究开发有限公司编著.药物临床信息参考.成都: 四川出版集团四川科学技术出版社,2006:1045. 【21】 章思规等编著.精细化学品及中间体手册:上卷.北京:化学工业出版社,2004:339. 【22】 Horning E C,Edutor-in-Chief.Organic Syntheses Collective:Volum 3.New York;John Wiley,1955:197-199,811-813. 【23】 刘少诚等.中国药物化学杂志,1992,2:56-57. 【24】 岑均达.中国医药工业杂志,1997,28:197-199. 【25】 周学良主编.精细化工产品手册: 药物. 北京: 化学工业出版社,2003:638-640.
备注
暂无
表征图谱

暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号