接近遗传密码符号扩增的生物合成方法:非自然DNA碱基对的分子设计
- 2016-01-22
- PC
1.简介
2010年, Craig Venter's 团队报道了一种新兴的含有一个人工合成具有1.08 M 碱基基因组的支原体细菌。1这个成就是化学家和生物学家协作努力通过五年研究出来的结果。初始细胞的生成需要很大的努力,一旦制得人工细胞,他们就会像自然细胞一样增殖。因此,人工设计的细胞可以合理的成本再生,用作合成有用蛋白质和其他材料的生物工厂。现有的生物系统的重复设计是一个合成生物学途径的例子。2
另一合成生物学方法的类型是构建一个新的生物系统,用最新的生物组分来建造。在这个方法中,新的人造组分用于开发服务于某个目标,在生物系统中他们与侧边的自然组分一起运作。新的组分由重复的“proof of concept”试验创建。原型组分基于某个概念或者主意设计,并根据物理和生物实验结果进行改进。在这里,我们通过对DNA遗传密码符号扩展的非自然碱基对的创建,描述这种类型的生物合成方法。3-8
2. 非自然碱基对的发展
地球生命遗传信息在DNA序列中有四个不同碱基对编码,腺嘌呤(A),鸟嘌呤(G), 胞嘧啶(C)和胸腺嘧啶(T)。DNA双链分子中,A选择性的与T配对,G与C配对。碱基配对原则是基本的通过复制、转录和翻译的遗传信息流。因此,DNA中引入一个人为创建的碱基对可以增加遗传字母,扩大遗传信息,提供一个新的生物技术能够与特定的新组分结合为核酸和蛋白质(Figure 1)。9此外,最近的利用人工碱基在复制和转录方面的研究已经显示出新的分子相互作用和生物反应机理,仅仅通过自然组分利用生物分子标准进行馋鬼分子观测不出来。而且,人为的提高生物系统遗传密码符号对于化学家来说是一个巨大的挑战。
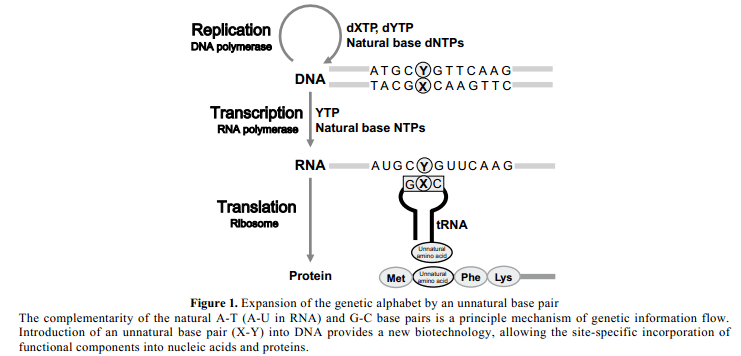
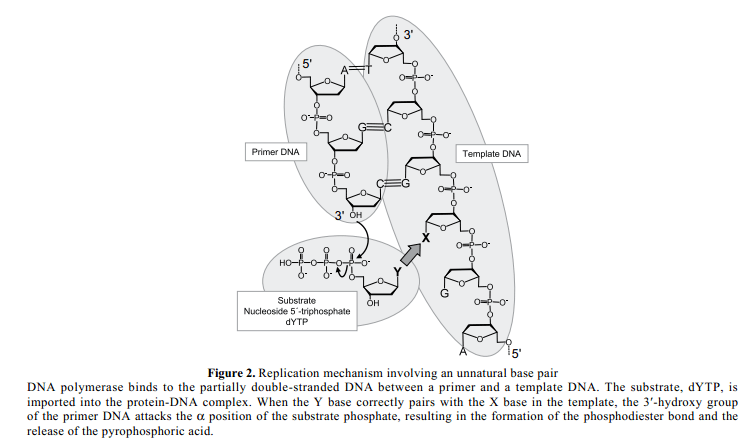
最重要的问题是,费自然碱基对(X-Y) 作为三分之一的碱基对具有很高的特有的选择性,也就是说,X选择性与Y配对,与此同时, A-T和G-C在生物系统中配对(Figure 2)。在复制中,DNA聚合酶结合底物与模板链之间部分双链DNA片段。随后,5'-三磷酸何干(dNTP, substrate)进入DNA聚合酶络合物。当基质在模板中与正确的伙伴配对,然后引物3'-羟基基团的氧原子进攻基板三磷酸α 位置。这个结果导致在引物和导入物之间形成磷酸双酯键,并且释放出焦磷酸做为一个离去集团。引物结合正确的基质后,DNA聚合酶滑向模板DNA 链,下一个基质结合发生。复制中自然碱基配对的选择性非常的高。例如,大肠杆菌DNA聚合酶I的克列诺片段,10,000 次结合会出现一次不正确的结合,10也就是说克列诺片段每一次复制的自然碱基对选择性为99.99%。
3. 自然碱基对: A-T和G-C
创建一个非自然碱基对,我们需要了解为什么A-T 和G-C 碱基对按照他们的化学、物理、生物状态在复制中有如此高的选择性。在A-T和G-C 配对中,嘌呤碱基(A or G)与嘧啶碱基(T or C)配对, 两个氢键用于A-T,三个用于G-C (Figure 3)。因此,每一个碱基对糖苷键的距离约为10.7-11.0 Å,并且双螺旋DNA 分子具有规则的人字齿轮结构。氢键在质子予体残基和质子受体原子或残基中间形成。A-T和G-C对之间,氢键模式各不相同,因此,A 与T, G 和C配对总是一种常规的螺旋结构。

然而,最近的研究已经表明, 在复制中氢键的形成没有必要搭配正确的碱基对。
1995年,Eric Kool 和他的同事们设计合成了一个疏水性非自然Z–F对,其中Z和F 的形状分别模仿A 和T(Figure 4)。11-13 对于Z,A 中1- 和3-氮被碳替代,6-氨基被甲基替代。对于F,T中3-氨基被C-H替代,2-和4-酮基由氟原子替代。通常,一个氟原子质子受体的能力低于酮十倍。Kool团队化学方法合成特定位置中含有Z 或F的 DNA 模板 ,以及他们的核苷三磷酸盐(dZTP和dFTP),作为底物在体外进行复制实验。他们表明,大肠杆菌DNA聚合酶I的克列诺片段在模板中有效的结合了dFTP 和dTTP 成为DNA ,对立与Z 或A,也与dZTP 、dATP 对立与F或T合并。相比之下,dCTP 相对Z 和 dGTP相对F的合并效率较低。因此,疏水性非自然Z-F对在复制中作用,与A-T对兼容,这表明,复制中配对碱基之间形状互补性重要于氢键互补性。14
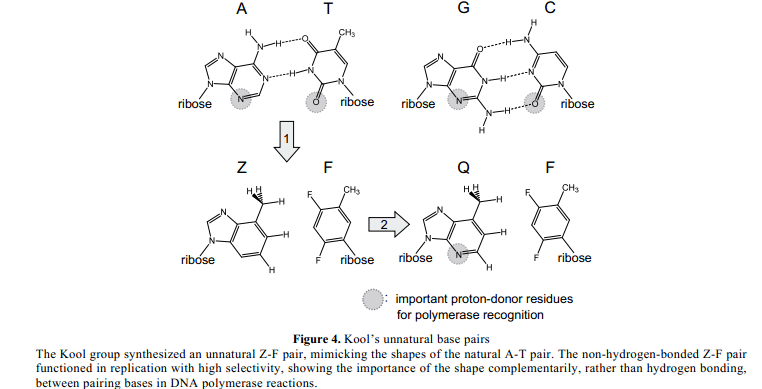
通过聚合酶来识别,A 和G中的3-氮, C和T中的2-酮作为氢键受体对于小沟是很必要的,在聚合酶中与具体的氨基酸残基相互作用(Figure 3)。15例如,缺失2-酮基的嘧啶类似物基质在PCR中不能够并入DNA ( P olymerase C hain R eaction).16 Kool的Z碱基相对于嘌呤3-氮缺少氢键受体原子,因此,他们新近研发Q 碱基,其位置上有一个氮(Figure 4)。17克列诺片段被复制的Q-F碱基对比Z- F对更高效。其他的因素,比如基质疏水性,基质偶极距,相邻级之间堆积相互作用,以及CH- π相互作用18,在复制中对于碱基对选择性也很重要。
考虑到这些,目前为止非自然碱基对的各种类型已经发展至此。下面的季节中么,我们将介绍具有代表性的非自然碱基对以及他们的发展进程。
4.Benner团队的亲水性非自然碱基对
在1980年代后期,Steven Benner和他的同事们报道了四种类型的具有不同氢键几何体的 非自然碱基对,这不同于A-T和G-C对。19, 20 其中一个典型代表就是6-氨基-2-ketopurine (isoG)和2-氨基-4-ketopyrimidine (isoC)间的碱基对,他们分别是G和C的结构异构体(Figure 5)。19Benner团队证实了isoG-isoC对通过克列诺片段在复制中互补作用。此外,isoG基质(isoGTP)并入RNA 相对于isoC 并入DNA 模板,通过T7 RNA聚合酶。21 此外,1992年,isoG-isoC对用于体外翻译系统,通过一个短的化学合成mRNA(含isoC)和一个3-iodotyrosyl tRNA (含isoG) ,使得不标准氨基酸特定合并成为多肽。22
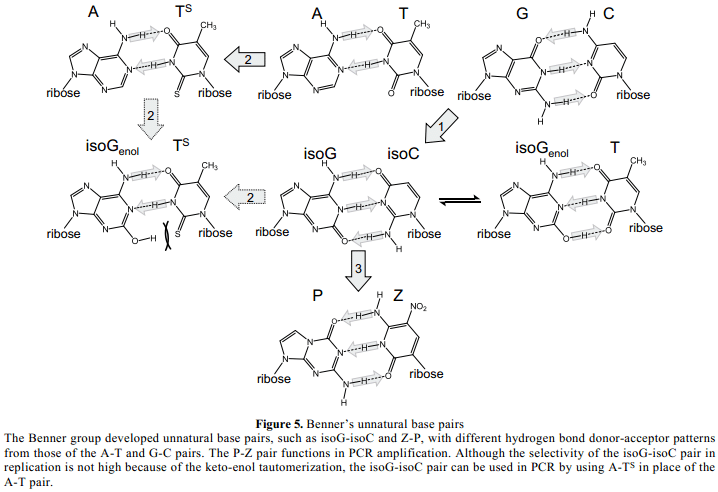
Benner团队这些开创性的研究对利用非自然碱基对的遗传膨胀系数受到了很大的关注。然而,这些非自然碱基对仍有很多的缺点。比如,isoG碱基溶液中经受keto-enol互变异构化,isoG碱基与T烯醇化(Figure 5)。21 除了isoG的问题,isoC中的2-氨基,相当于自然碱基的2-keto位置,降低与聚合酶的相互作用。21此外,isoC的核苷在水溶液中不稳定,室温下,四天以后水中中性环境下会有50% 的isoC核苷三磷酸分解掉。23尽管在isoC碱基位置3处(相当于自然嘧啶碱基位置5处)引入甲基可以改良其稳定性,然而 甲基-isoC 核糖核苷仍然易受其β-糖苷键差向异构化作用,从β-转换为α-形式。最近,Benner团队通过在位置3处引入一个硝基解决了这个问题,24 如下所示。
2005年, Benner团队通过利用一个改性的T 碱基,2-thiothymine (TS),替代T (Figure 5), 解决了isoG keto-烯醇互变异构化的难题,用于isoG-isoC碱基对系统。25 TS中大尺寸的硫原子阻止了isoG形式中与烯醇的配对, 但仍是没有空间斥力的配对。这个策略类似于Kool的配对碱基键形状适合概念,以及Harry 1988年报道的的非自然碱基对的概念,26 他利用一个改良的鸟嘌呤,6-thioguanine作为一个新的碱基。非自然isoG-isoC对与A-TS对在酶聚合链反应中共同作用:isoG-isoC对的每一次复制选择性达到98%,而isoG-isoC选择性在没有TS的帮助下每次复制达到93%。25然而,98%非自然碱基对选择性对于复制来说是不够的,并且非自然碱基对的保留率在PCR反应扩大DNA 片段的20周期中变为67% (0.9820 =0.67)。因此,对于复制中非自然碱基对的实际使用中,每一次复制超过99% 的选择性是必要的。
2007年,Benner团队改进了非自然碱基对系统,并研发了P-Z 对,其在PCR扩增中不需使用A-TS对(Figure 5)。24 不同于isoG,P碱基在溶液中不经历不良的互变异构化。Z碱基中的硝基基团由于其亚氨基的去质子化,阻止了它的核苷衍生物的差向异构化。虽然P-Z对选择性当时在PCR中为97.5% ,但是最近的报道表明优化PCR 条件下改良的选择性在99.8%。27
5. Romesberg团队的疏水性非自然碱基对
Kool的关于非氢键碱基对的开创性实验引发疏水性碱基对作为非自然碱基对的候选物。Floyd Romesberg和他的同事开发了大量的疏水性非自然碱基对,在复制系统中检测他们的能力。1999年,他们报道了一个疏水性异喹啉衍生物PICS(Figure 6),其自我互补碱基对在双螺旋DNA 片段中具有很高的热稳定性。28此外,PICS酶作用物与DNA酶结合通过克列诺片段与PICS在模板中相对应。
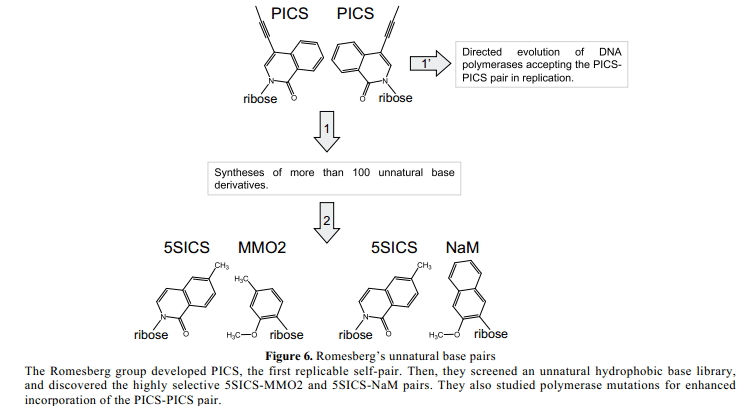
不幸的是,在PICS 基板中参入PICS后,引物暂停延伸。这是因为PICS-PICS对的形状对于常规DNA双螺旋的空间来说太大了,其PICS 碱基相互堆叠,不允许聚合酶识别。因此,他们详尽的设计和合成了另外的疏水性非自然碱基对,并在复制中检验了它们的能力。29-35
2009年,他们成功的开发了疏水性非自然碱基对5SCIS-MMO2和5SCIS-NaM (Figure 6),他们的功能是作PCR扩增中三分之一的碱基对。36, 37与之前的PICS–PICS对相比, 这些碱基对的空间互补形状有所改进。5SCIS–NaM 对在PCR中最佳的保真度达到99.8%,尽管它取决于周围非自然碱基对的排列顺序。这些疏水性碱基对通过T7 RNA 聚合酶在转录中也起作用。38 该团队还通过一个进化工程技术设计出DNA聚合酶,通过采用突变聚合酶了解决复制中使用PICS-PICS 碱基对时聚合酶暂定问题。39
6. Hirao团队的一系列非自然碱基对
我们团队在1997年之后也研究了非自然碱基对。大量的“proof of concept”实验后,我们于201年开发出第一个非自然碱基,在2-氨基-6-dimethylaminopurine (x)和2-oxopyridine (y)之间(Figure 7)。40这个x-y设计包含两个概念:Benner氢键模式(它不同于自然碱基对’s)和一个位阻效应。这个x-y对具有两个氢键,x 的2-aminopurine 部分通过一个类似的氢键作用与T 配对。因此,我们在x的6位置处添加一个巨大的二甲胺基团, 通过T中6-dimethylamino 基团和4-keto基团之间的空间位阻来排除x-T配对。与 T相反, y 具有一个氢键代替了keto 基。x-y 对功能在转录中具有很高的选择性,T7RNA聚合酶混合y三磷酸基质(yTP) 成为RNA,相对于x 在DNA模板中,具有高于95%的选择性。
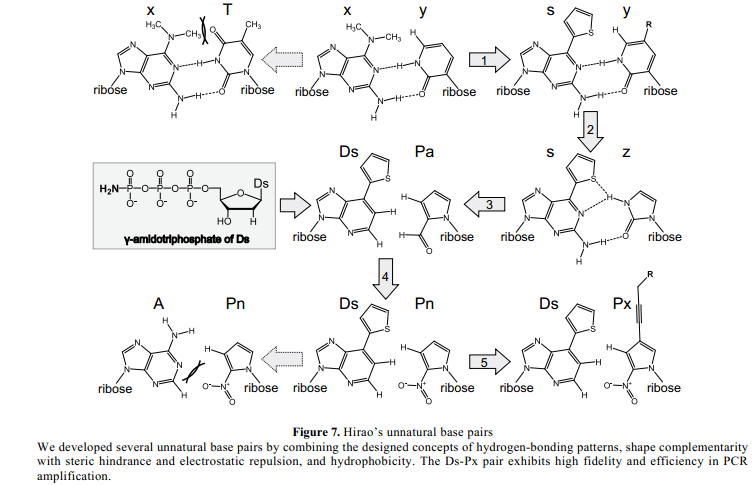
此外,我们通过用一个噻吩基替代二甲胺基改进了非自然碱基对的形状互补性。因此,我们开发了2-amino-6-thienylpurine (s)作为y的配对搭档 (Figure 7)。41噻吩基的平面化比X二甲胺基更加有效的排除了与T 的错误配对。此外,平面性增加了s 碱基与相邻碱基在DNA 链中叠加的能力。相比x-y 对,s-y对在转录中的选择性和效率都有了很大的改进 不仅仅yTP 也包括yTPs的一些部分,通过T7 RNA聚合酶,比如biotin-和fluorophore-linked y 碱基,与RNA结合,相对应于DNA模板中。42-442002年,通过结合特定的含有s–y对和一个E. coli体外翻译系统的转录,我们成功的特定结合一个非标准氨基酸,3-chlorotyrosine变为一个蛋白质。41 因此,s–y对可疑作为体外翻译和转录系统中第三个碱基 。
我们试图进一步的改进我们这些非自然碱基对。一个主要的障碍是s–y对复制中不足的选择性。PCR的一个DNA含有s–y碱基对片段10个周期过后,大约有40%的非自然碱基对被自然碱基对所替代。每一次复制中s–y对选择性是~95%(40%的替代,10周期PCR过后: 1−0.9510=~0.4),因此,对于非自然碱基对复制中超出99% 的选择性时必须的,一个非自然碱基对约~90% (0.9910 =~0.90)可保留。因此,我们严格的精炼碱基对之间的形状互补,并设计了一个五元环核苷模型,imidazoline-2-one (z),45 代替六元环y,作为s的配对搭档 (Figure 7)。此外,我们祛除了来自s-z对之间的含有氢键相互作用的原子和残基,46 在7-(2-thienyl)imidazo[4,5- b]-pyridine (Ds)和pyrrole-2-carbaldehyde (Pa)之间开发了一个疏水性非自然碱基对 (Figure 7, 参阅化学合成附录)。47我们在Pa 碱基上添加一个醛基,作为一个质子受体用于聚合酶之间的相互作用。
Ds–Pa对的问题是self-complementary Ds-Ds配对在复制中也有竞争力的发生。也发现了Romesberg’s PICS–PICS对,在Ds合并含有 对立面的Ds时的扩展停顿。幸运的是,我们发现了一个改良过后的Ds三磷酸基质,γ-amidotriphosphate, 选择性的结合opposite Pa,但是结合opposite Ds之后显示出大幅度的减少。47改进过后的基质合并,因为基质的amidopyrophosphate部分被祛除作为一个聚合作用中的离去集团,DNA具有母磷酸二酯键合。为了PCR扩增,我们除了Ds也使用A中的γ –amidotriphosphate,来降低dATP 相对Pa.的插入性错误。2006年,我们使用A中的γ –amidotriphosphate和Ds,成功的完成了含有Ds–Pa对的高选择性PCR扩增,其中每一次复制Ds–Pa对的选择性达到99%以上。一个偶然的失误,我们通过处理三磷酸合成中间体意外的合成了γ -amidotriphosphates,5'-三磷酸酯环状物,具有浓缩氨。
我们进一步的改进了Ds-Pa对,用硝基替代Pa中的醛基,因此设计了2-nitropyrrole (Pn)(Figure 7)。48 Pn的硝基通过硝基中的氧和A中的1-氮之间的静电斥力可以减少A opposite Pa的插入性错误。另外,我们在Pn碱基4-位置处添加丙炔基,开发出 2-硝基-4-propynylpyrrole (Px) 作为Ds的配对搭档(Figure 7)。49 丙炔基增加了疏水性,增强了聚合酶之间的相互作用。因此,Ds-Px对在PCR中的功能不需要γ –amidotriphosphates的援助。含有Ds-Px对的DNA 片段通过PCR 40周期后被放大108-fold,超过97%的Ds–Px对仍被保留在扩大的DNA中。The selectivity of the Ds–Px对的选择性在每次复制中达到99.9%以上,这是迄今为止非自然碱基对中最高的选择性。50
7. 结论
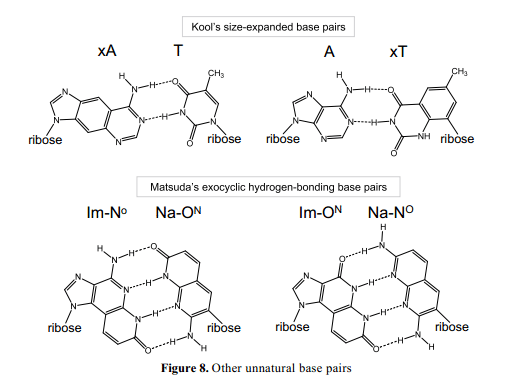
在过去的20年中,非自然碱基对合成生物学有何快速的发展,各种非自然碱基对被研发出来(Figure 8)。4, 51-54 这些非自然碱基对被应用在DNA和RNA分子特定的荧光标记,42, 55 固定,43, 47 特定检测,56-60 和结构分析61-63中。虽然非自然碱基对在翻译中的应用程序还在开发研制,22, 41但到蛋白质合成技术开发应用于非标准氨基酸特定结合为蛋白质这只是一个时间问题。先前的非自然碱基对的研究仅限于体外实验。现在我们又几种类型的非自然碱基对有可能用于体内实验。将来,利用非自然碱基对扩增遗传密码符号竟会应用到细胞,比如Venter's artificial cell, 有效的产生人造蛋白质和细胞中跟踪表达目标基因。我们期望这一领域对新生代生物技术有进一步的发展。


References
1. D. G. Gibson, J. I. Glass, C. Lartigue, V. N. Noskov, R.Y. Chuang, M. A. Algire, G. A. Benders, M.G. Montague, L. Ma, M. M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E. A. Denisova, L. Young, Z. Q. Qi, T. H. Segall-Shapiro, C. H. Calvey, P. P. Parmar, C. A. Hutchison, 3rd, H. O. Smith, J. C. Venter, Science 2010, 329, 52–56.
2. D. Sprinzak, M. B. Elowitz, Nature 2005, 438, 443–448.
3. D. E. Bergstrom, Curr. Protoc. Nucleic Acid Chem. 2009, Chapter 1, Unit 1 4.
4. A. T. Krueger, E. T. Kool, Chem. Biol. 2009, 16 , 242–248.
5. I. Hirao, Curr. Opin. Chem. Biol. 2006, 10 , 622–627.
6. A. A. Henry , F. E. Romesberg, Curr. Opin. Chem. Biol. 2003, 7, 727–733.
7. S. A. Benner, A. M. Sismour, Nat. Rev. Genet. 2005, 6, 533–543.
8. I. Hirao, M. Kimoto, R. Yamashige, Acc. Chem. Res . in press.
9. R. F. Service, Science 2000, 289, 232–235.
10. T. A. Kunkel, K. Bebenek, Annu. Rev. Biochem. 2000, 69 , 497–529.
11. S. Moran, R. X. Ren, E. T. Kool, Proc. Natl. Acad. Sci. USA 1997, 94 , 10506–10511.
12. J. C. Morales, E. T. Kool, Nat. Struct. Biol. 1998, 5, 950–954.
13. K. M. Guckian, T. R. Krugh, E. T. Kool, Nat. Struct. Biol. 1998, 5, 954–959.
14. T. J. Matray, E. T. Kool, Nature 1999, 399, 704–708.
15. E. T. Kool, Annu. Rev. Biochem. 2002, 71 , 191–219.
16. M. J. Guo, S. Hildbrand, C. J. Leumann, L. W. McLaughlin, M. J. Waring, Nucleic Acids Res. 1998, 26 , 1863–1869.
17. J. C. Morales, E. T. Kool, J. Am. Chem. Soc. 1999, 121, 2323–2324.
18. O. Takahashi, Y. Kohno, M. Nishio, Chem. Rev. 2010, 110, 6049–6076.
19. C. Switzer, S. E. Moroney, S. A. Benner, J. Am. Chem. Soc. 1989, 111, 8322–8323.
20. J. A. Piccirilli, T. Krauch, S. E. Moroney, S. A. Benner, Nature 1990, 343, 33–37.
21. C. Y. Switzer, S. E. Moroney, S. A. Benner, Biochemistry 1993, 32 , 10489–10496.
22. J. D. Bain, C. Switzer, A. R. Chamberlin, S. A. Benner, Nature 1992, 356, 537–539.
23. J. J. Coegel, S. A. Benner, Helv. Chim. Acta 1996, 79 , 1863–1880.
24. Z. Yang, A. M. Sismour, P. Sheng, N. L. Puskar, S. A. Benner, Nucleic Acids Res. 2007, 35 , 4238–4249.
25. A. M. Sismour, S. A. Benner, Nucleic Acids Res. 2005, 33 , 5640–5646.
26. H. P. Rappaport, Nucleic Acids Res. 1988, 16 , 7253–7267.
27. Z. Yang, F. Chen, J. B. Alvarado, S. A. Benner, J. Am. Chem. Soc. 2011, 133, 15105–15112.
28. D. L. McMinn, A. K. Ogawa, Y. Wu, J. Liu, P. G. Schultz, F. E. Romesberg, J. Am. Chem. Soc. 1999 , 121 , 11585–11586.
29. E. L. Tae, Y. Wu, G. Xia, P. G. Schultz, F. E. Romesberg, J. Am. Chem. Soc. 2001, 123, 7439–7440.
30. A.A. Henry, C. Yu, F. E. Romesberg, J. Am. Chem. Soc. 2003, 125, 9638–9646.
31. A. A. Henry, A. G. Olsen, S. Matsuda, C. Yu, B. H. Geierstanger, F. E. Romesberg, J. Am. Chem. Soc. 2004, 126, 6923–6931.
32. A. M. Leconte, S. Matsuda, G. T. Hwang and F. E. Romesberg, Angew. Chem. Intl. Ed. Engl. 2006, 45 , 4326–4329.
33. S. Matsuda, J. D. Fillo, A. A. Henry, P. Rai, S. J. Wilkens, T. J. Dwyer, B. H. Geierstanger, D. E. Wemmer, P. G. Schultz, G. Spraggon, F. E. Romesberg, J. Am. Chem. Soc. 2007, 129, 10466–10473.
34. A. M. Leconte, G. T. Hwang, S. Matsuda, P. Capek, Y. Hari, F. E. Romesberg, J. Am. Chem. Soc. 2008, 130, 2336–2343.
35. Y. J. Seo, G. T. Hwang, P. Ordoukhanian, F. E. Romesberg, J. Am. Chem. Soc. 2009, 131, 3246–3252.
36. D. A. Malyshev, Y. J. Seo, P. Ordoukhanian, F. E. Romesberg, J. Am. Chem. Soc. 2009, 131, 14620–14621.
37. D. A. Malyshev, D. A. Pfaff, S. I. Ippoliti, G. T. Hwang, T. J. Dwyer, F. E. Romesberg, Chemistry 2010, 16 , 12650–12659.
38. Y. J. Seo, S. Matsuda, F. E. Romesberg, J. Am. Chem. Soc. 2009, 131, 5046–5047.
39. A. M. Leconte, L. Chen, F. E. Romesberg, J. Am. Chem. Soc. 2005, 127, 12470–12471.
40. T. Ohtsuki, M. Kimoto, M. Ishikawa, T. Mitsui, I. Hirao, S. Yokoyama, Proc. Natl. Acad. Sci. USA 2001, 98 , 4922–4925.
41. I. Hirao, T. Ohtsuki, T. Fujiwara, T. Mitsui, T. Yokogawa, T. Okuni, H. Nakayama, K. Takio, T. Yabuki, T. Kigawa, K. Kodama, K. Nishikawa, S. Yokoyama, Nat. Biotechnol. 2002, 20 , 177–182.
42. R. Kawai, M. Kimoto, S. Ikeda, T. Mitsui, M. Endo, S. Yokoyama, I. Hirao, J. Am. Chem. Soc. 2005, 127, 17286–17295.
43. K. Moriyama, M. Kimoto, T. Mitsui, S. Yokoyama, I. Hirao, Nucleic Acids Res. 2005, 33 , e129.
44. I. Hirao, Biotechniques 2006, 40, 711–715.
45. I. Hirao, Y. Harada, M. Kimoto, T. Mitsui, T. Fujiwara, S. Yokoyama, J. Am. Chem. Soc. 2004, 126, 13298–13305.
46. T. Mitsui, A. Kitamura, M. Kimoto, T. To, A. Sato, I. Hirao, S. Yokoyama, J. Am. Chem. Soc. 2003, 125, 5298–5307.
47. I. Hirao, M. Kimoto, T. Mitsui, T. Fujiwara, R. Kawai, A. Sato, Y. Harada, S. Yokoyama, Nat. Methods 2006, 3, 729–735.
48. I. Hirao, T. Mitsui, M. Kimoto, S. Yokoyama, J. Am. Chem. Soc. 2007, 129, 15549–15555.
49. M. Kimoto, R. Kawai, T. Mitsui, S. Yokoyama, I. Hirao, Nucleic Acids Res. 2009, 37 , e14.
50. R. Yamashige, M. Kimoto, Y. Takezawa, A. Sato, T. Mitsui, S. Yokoyama, I. Hirao, Nucleic Acids Res. in press.
51. J. Gao, H. Liu and E. T. Kool, J. Am. Chem. Soc. 2004, 126, 11826–11831.
52. N. Minakawa, S. Ogata, M. Takahashi, A. Matsuda, J. Am. Chem. Soc. 2009, 131, 1644–1645.
53. S. Hikishima, N. Minakawa, K. Kuramoto, Y. Fujisawa, M. Ogawa, A. Matsuda, Angew. Chem. Int. Ed. Engl. 2005, 44 , 596–598.
54. C. Kaul, M. Muller, M. Wagner, S. Schneider, T. Carell, Nat. Chem. 2011, 3, 794–800.
55. M. Kimoto, T. Mitsui, S. Yokoyama, I. Hirao, J. Am. Chem. Soc. 2010, 132, 4988–4989.
56. C. B. Sherrill, D. J. Marshall, M. J. Moser, C. A. Larsen, L. Daude-Snow, S. Jurczyk, G. Shapiro, J. R. Prudent, J. Am. Chem. Soc. 2004, 126, 4550–4556.
57. S. C. Johnson, D. J. Marshall, G. Harms, C. M. Miller, C. B. Sherrill, E. L. Beaty, S. A. Lederer, E. B. Roesch, G. Madsen, G. L. Hoffman, R. H. Laessig, G. J. Kopish, M. W. Baker, S. A. Benner, P. M. Farrell, J. R. Prudent, Clin. Chem. 2004, 50 , 2019–2027.
58. P. Sheng, Z. Yang, Y. Kim, Y. Wu, W. Tan, S. A. Benner, Chem. Commun. 2008, 5128–5130.
59. S. Hoshika, F. Chen, N. A. Leal, S. A. Benner, Angew. Chem. Int. Ed. Engl. 2010, 49 , 5554–5557.
60. R. Yamashige, M. Kimoto, T. Mitsui, S. Yokoyama, I. Hirao, Org. Biomol. Chem. 2011, 9, 7504–7509.
61. M. Kimoto, M. Endo, T. Mitsui, T. Okuni, I. Hirao, S. Yokoyama, Chem. Biol. 2004, 11 , 47–55.
62. M. Kimoto, T. Mitsui, Y. Harada, A. Sato, S. Yokoyama, I. Hirao, Nucleic Acids Res. 2007, 35 , 5360–5369.
63. Y. Hikida, M. Kimoto, S. Yokoyama, I. Hirao, Nat. Protoc. 2010, 5, 1312–1323.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号