
结构式
| 物竞编号 | 0004 |
|---|---|
| 分子式 | Pd |
| 分子量 | 106.4 |
| 标签 | 纯元素, 催化剂, 电子高纯单质原料及中间体 |
编号系统
CAS号:7440-05-3
MDL号:MFCD00011167
EINECS号:231-115-6
RTECS号:RT3480500
BRN号:暂无
PubChem号:暂无
物性数据
1. 性状:银白色金属。(面心立方结晶)。延展性强。
2. 密度(g/mL,20℃):12.02
3. 相对蒸汽密度(g/mL,空气=1):未确定
4. 熔点(ºC):1554
5. 沸点(ºC,常压):2970
6. 沸点(ºC,5.2kPa):未确定
7. 折射率:未确定
8. 闪点(ºC):未确定
9. 比旋光度(º):未确定
10. 自燃点或引燃温度(ºC):未确定
11. 蒸气压(mmHg,25ºC):未确定
12. 饱和蒸气压(kPa, ºC):未确定
13. 燃烧热(KJ/mol):未确定
14. 临界温度(ºC):未确定
15. 临界压力(KPa):未确定
16. 油水(辛醇/水)分配系数的对数值:未确定
17. 爆炸上限(%,V/V):未确定
18. 爆炸下限(%,V/V):未确定
19. 溶解性:溶于王水、热硝酸、硫酸,微溶于盐酸,不溶于冷水和热水。
毒理学数据
1、其它多剂量毒性:大鼠经口TDLo:9100mg/kg/26W-I,对眼睛和皮肤有刺激作用。
生态学数据
该物质对环境有危害,应特别注意对水体的污染。
分子结构数据
1、摩尔折射率:无可用的
2、摩尔体积(cm3/mol):无可用的
3、等张比容(90.2K):无可用的
4、表面张力(dyne/cm):无可用的
5、介电常数:无可用的
6、极化率(10-24cm3):无可用的
7、单一同位素质量:105.903483 Da
8、标称质量:106 Da
9、平均质量:106.42 Da
计算化学数据
1、 疏水参数计算参考值(XlogP):
2、 氢键供体数量:0
3、 氢键受体数量:0
4、 可旋转化学键数量:0
5、 互变异构体数量:
6、 拓扑分子极性表面积(TPSA):0
7、 重原子数量:1
8、 表面电荷:0
9、 复杂度:0
10、 同位素原子数量:0
11、 确定原子立构中心数量:0
12、 不确定原子立构中心数量:0
13、 确定化学键立构中心数量:0
14、 不确定化学键立构中心数量:0
15、 共价键单元数量:1
性质与稳定性
1.避免与氧化物,强酸,碱,卤素接触。溶于王水、 热硝酸、硫酸,微溶于盐酸,不溶于冷水和热水。超细钯粉:灰黑色粉末。微米级粒度的钯粉,有鳞片状、无定形和球状。鳞片状钯粉的松装密度0.67~0.95g/cm3。摇实密度1.6~2.00g/cm3。平均粒度0.35~0.48μm,比表面积3.3~5.2m2/g。电性能稳定,对氢吸附能力大。其粉体遇高温、明火能燃烧。与甲酸或四氢硼酸钠反应放出氢气。与异丙醇发生剧烈反应。
在空气中加热至800℃,生成暗淡无光的氧化膜,高于此温度即分解。能大量吸附多种气体,尤其是H2。室温下能抗氢氟酸、磷酸、盐酸和硫酸的侵蚀。溶于王水、热HNO3、H2SO4。微溶于盐酸。
贮存方法
储存于阴凉、通风的库房。远离火种、热源。应与酸类、卤素分开存放,切忌混储。配备相应品种和数量的消防器材。储区应备有合适的材料收容泄漏物。应贮存在干燥、清洁的库房内。
合成方法
1.工业生产可从矿石用干法制造;亦可以铜、镍的硫化矿制取铜、镍的生产过程中生成的副产物作为原料,用湿法冶炼制得。
湿法把已提取镍、铜后的残留组分作为原料,加入王水进行抽提,过滤,向滤液中加入氨和盐酸进行反应,生成氯钯酸铵沉淀。经精炼,过滤,把氯钯酸铵用氢气还原,制得约99.95%钯成品。
超细钯粉制法:化学处理金属钯制取球形超细钯粉。
2.在PdCl2或Na2PdCl4中加入NH4Cl使可能存在的微量Pt生成(NH4)2PtCl6沉淀。滤液中加入过量氨水并煮沸,必要时再次过滤。然后用盐酸酸化,生成很纯的黄色[PdCl2(NH3)2]沉淀。若所得沉淀呈暗浊的黄色,其中即含有少量不溶于冷氨水的[RhCl(NH3)5]Cl2。可将沉淀与氨水共煮,取出滤液,再次用盐酸酸化,可得亮黄色的很纯的[PdCl2(NH3)2]晶体。将晶体在H2气流中灼烧还原,得到浅灰色的海绵钯。
3.工业生产可以矿石用干法制造;亦可以铜、镍的硫化矿制取铜、镍的生产过程中生成的副产物作为原料,用湿法冶炼制得。
湿法 将已提取镍、铜后的残留组分作为原料,加入王水进行抽提、过滤,向滤液中加入氨和盐酸进行反应,生成氯化钯酸铵沉淀。经精炼、过滤,将氯化钯酸铵用氢气还原,制得约99.95%的钯成品。
4.在0.443克(2 .5摩尔)氯化把溶于40毫升绝对甲醇(或其它溶剂)的溶液中,在搅拌和室温下,5~10分钟内分次加入0.19克(5.0毫摩尔)粉状氢化硼钠。继续搅拌20分钟,或直到不再放出气体为止。搅拌停止后,黑色的反应产物很快就沉降下来用倾倒并洗涤的办法更换溶剂两次。此催化剂可以直接使用,或贮放在有塞瓶中的液体下面。
用途
1.用于电器仪表、精密合金等。电气仪表,化学工业及制造精密合金等工业用。主要用于制催化剂,还用于制造牙科材料、手表和外科器具等。用于制合金、钟表轴承及零件、镜面、加氢催化剂等。超细钯粉用途:产品主要用于制作厚膜导体浆料及净化材料。
2.商品化的钯催化剂本篇介绍三种:①钯-硫酸钡催化剂,也叫罗森蒙德催化剂;②钯-碳催化剂,黑色粉末或者是含有0.5%~30%钯的小球,不溶于所有的有机溶剂和酸性溶液;③钯-碳酸钙-乙酸铅催化剂,异相催化剂,不溶于水合有机溶剂。
①钯-硫酸钡催化剂
钯催化的将酰氯转变为醛的氢化反应,也称为“罗森蒙德反应” (式1)[1]。该反应初期的方法,都是采用将氢气通入到酰氯的二甲苯或甲苯溶液中来完成的。尽管这一方法能够适用于大部分酰氯化合物,但是进一步的还原反应如将醛转变为醇,以及进一步形成酯、醚和烃类化合物等副反应会严重影响罗森蒙德反应的产率。
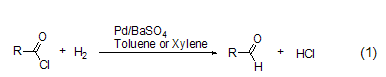
后续研究发现了一种更为可行的操作方法,即在二甲苯或甲苯溶剂中,采用钯/硫酸钡作催化剂,采用喹啉-硫作调节剂,通入氢气后回流反应能够高产率地实现萘酰氯的氢化反应 (式2)[2]。
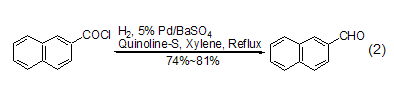
为了进一步克服传统罗森蒙德反应中使用的高温以及通氢气的危险,将钯/硫酸钡替换为钯/碳,并加入无水乙酸钠作为盐酸的吸收剂,能够在温和条件下实现密闭体系的罗森蒙德反应 (式3)[3],从而为其工业化应用打下了良好的基础。
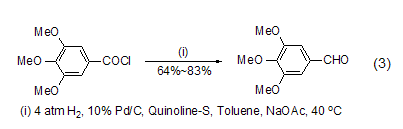
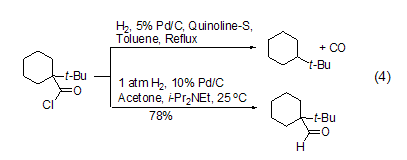
在反应体系中加入盐酸吸收剂如N,N-二甲基乙酰胺、乙酸钠和乙基二异丙基胺等都能有效促进反应的进行,能够在温和条件下实现立体禁阻的酰氯底物的氢化反应,如采用传统罗森蒙德反应方法,1-叔丁基环己酰氯的氢化反应得到的主要是叔丁基环己烷产物,而加入盐酸吸收剂乙基二异丙基胺后,能够高产率得到相应的醛化合物 (式4)[4]。
近年来的研究表明,不管是传统方法还是新方法,在实现酰氯到醛的转换中都是非常成功的,如采用传统方法,能够有效实现不饱和酰氯到不饱和醛的转换 (式5)[5]。
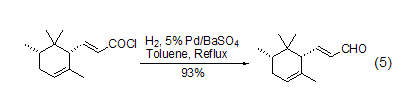
②钯-碳催化剂
Pd/C能够催化氢化烯烃、炔烃、酮、腈、亚胺、叠氮化物、硝基化合物、苯环以及杂环芳香化合物;可用于环丙烷、苯基衍生物、环氧化物、肼以及卤化物的氢解;同时也可以用于芳香化合物脱氢和醛的去酰基化反应。
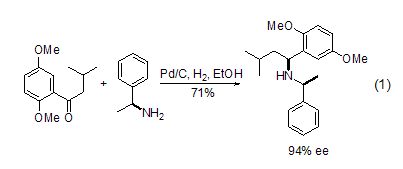
碳氮键 Pd/C在酸性或者铵溶液中,可以将腈氢化为一级胺。在没有酸性或者铵溶液时,将得到一级和二级胺的混合物。在酸性溶液中可以氢化得到醛或者醇。同腈一样,在酸性条件下,肟也能烷基化反应得到二级胺,用α-甲基苯胺作为手性辅助剂,可以实现对中间体亚胺的高度非对映异构选择性还原 (式1)[6]。这种方法提供了一种得到含氮杂环的很好途径。
Pd/C也能用于一些其它胺的前体,比如叠氮化物的还原烷基化反应。例如,呋喃糖环被开环后再关环,可以形成哌啶环 (式2)[7]。而吡喃基叠氮化合物可以发生类似的反应得到氮杂七元环。
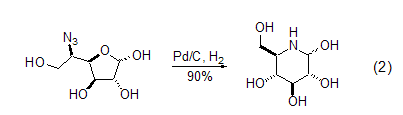
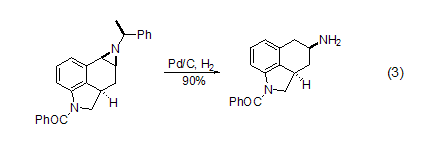
苯胺能被氢化得到较少烷基化的胺。碳氮键在转移氢化和常规氢化的条件下都能够断裂。烯丙胺也可以在Pd/C催化下发生去烯丙基化反应。1-氮杂环丙烷氢解可以得到开环胺,而且活性强的碳氮键能选择性地优先裂解 (式3)[8]。
碳卤键 芳香族卤化物(Cl, I, Br)可以用Pd/C催化氢化。反应中一般要用适量碱中和形成的酸。在没有酸中和剂的情况下,脱卤非常缓慢,甚至可能停止反应 (式4)[9]。
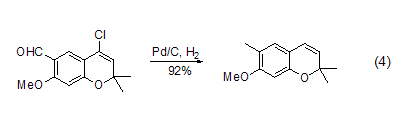
氮氮键 叠氮、重氮化合物可以在Pd/C作用下还原为胺。这些基团也可以看作是潜在的胺,发生氢化反应后再与分子内和对胺敏感的基团反应,得到不同的胺 (式5)[10]。
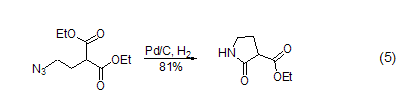
脱氢 在高温下,对碳环和杂环芳香化合物而言,Pd/C也是一种有效的脱氢试剂。烯酮可以转化为酚 (式6)[11]。
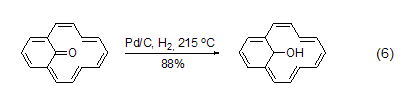
最近发现了一种新的复合材料——碳纳米纤维,由于碳纳米纤维有更大的比表面积,所以更适合作为金属催化剂的载体。在液相条件下,应用碳纳米纤维/钯催化剂,可以使四氢化萘发生脱氢化反应生成萘 (式7)[12]。
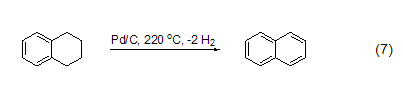
③钯-碳酸钙-乙酸铅催化剂
铅毒化的碳酸钙负载钯催化剂 (Lindlar催化剂)[13]是有机合成中最常用的异相氢化催化剂之一。Lindlar催化剂可以催化氢化还原多种有机官能团,例如:将芳环取代的硝基还原成为胺,但是与其它试剂相比较没有明显的特色。该试剂最独特的反应是高度选择性地将炔键还原至顺式双键。
炔键是有机化合物中最容易被还原的官能团之一,可以被部分还原生成烯烃,也可以彻底还原生成烷烃。如果被部分还原生成烯烃,可能得到顺式烯烃或者反式烯烃。对大多数已知的过渡金属催化剂来讲,不仅使产物停留在烯烃非常困难,而且缺乏立体选择性。Lindlar催化剂通过“毒化”和“去活”使其还原能力变得非常温和,高度选择性地将炔键还原至顺式双键。
使用Lindlar催化剂催化氢化炔键至双键的反应一般在室温和常压下进行,高纯氢气是最方便的氢源。反应一般在数分钟至数小时内完成,主要取决于底物的结构。反应的后处理非常简单,产物的产率一般很高或者接近定量。由于反应条件温和,该反应对其它官能团一般不产生明显的影响 (式1,式2)[14~17]。
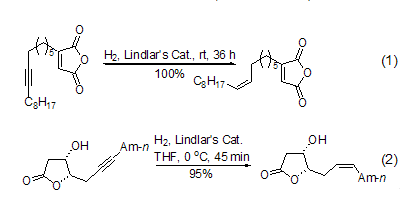
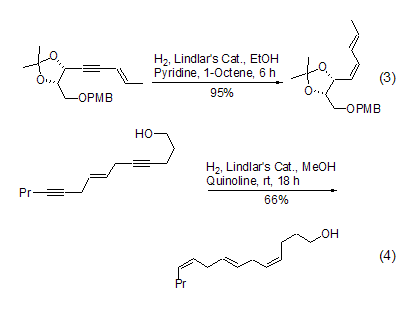
在反应体系中加入催化量的吡啶或者喹啉可以进一步使催化剂“去活”,增大生成烯烃的选择能力。在含有双键或者含有多个双键的分子中,高选择性和高产率地将炔键催化氢化至顺式双键最能够显示该试剂的优越性,特别适合复杂天然产物的合成 (式3)[18~20]。它可以将分子中多个炔键同时转化成为相应的顺式双键,使得复杂的合成变的异常简单 (式4)[21]。
在Lindlar催化剂的反应中,含有游离胺的炔烃化合物在选择性方面较差。如果在反应体系中加入适量的二亚乙基三胺,便可得到非常满意的结果 (式5)[22]。
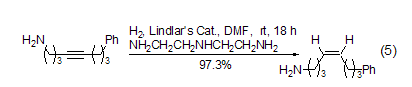
安全信息
危险运输编码:UN 3089 4.1/PG 2
危险品标志: 易燃
易燃  有毒
有毒  刺激
刺激
安全标识:暂无
危险标识:暂无
文献
1. Rylander, P. N. Hydrogenation Methods, Academic: New York, 1985. 2. Hershberg, E. B.; Cason, J. Org. Synth., Coll. Vol. 1985, 3, 551. 3. Wagner, D. P.; Gurien, H.; Rachlin, A. I. Ann. N. Y. Acad. Sci., 1970, 172, 186. 4. Peters, J. A.; van Bekkum, H. Recl. Trav. Chim. Pays-bas, 1971, 90, 1323. 5. Maurer, B.; Hauser, A.; Froidevaux, J.-C. Helv. Chim. Acta, 1989, 72, 1400. 6. Bringmann, G.; Kunkel, G.; Geuder, T. Synlett, 1990, 5, 253. 7. Dax, K.; Gaigg, B.; Grassberger, V.; Kolblinger, B.; Stutz, A. E. J. Carbohydr. Chem., 1990, 9, 479. 8. Martinelli, M. J.; Leanna, M. R.; Varie, D. L.; Peterson, B. C.; Kress, T. J.; Wepsiec, J. P.; Khau, V. V. Tetrahedron Lett., 1990, 31, 7579. 9. (a) Eszenyi, T.; Timar, T. Synth. Commun., 1990, 20, 3219. (b) Comins, D. L.; Weglarz, M. A. J. Org. Chem., 1991, 56, 2506. 10. (a) Lohray, B. B.; Ahuja, J. R. Chem. Comm., 1991, 95. (b) Lindstrom, K. J.; Crooks, S. L. Synth. Commun., 1990, 20, 2335. (c) Machinaga, N.; Kibayashi, C. Tetrahedron Lett., 1990, 31, 3537. (d) Chen, L.; Dumas, D. P.; Wong, C. H. J. Am. Chem. Soc., 1992, 114, 741. 11. Nelson, P. H.; Nelson, J. T. Synthesis, 1991, 192. 12. (a) Dung, T. P.; Morisaka, H.; Satoh, T.; Miura, M.; Nomura, M.; Matsui, H.; Yamaguchi, C.; Energy Fuels, 2003, 17, 658. (b) Dung, T. P.; Satoh, T.; Miura, M.; Nomura, M. Energy Fuels, 2005, 19, 731. 13. Lindlar, H. Helv. Chim. Acta, 1952, 35, 446. 14. Qin, D.; Byun, H.-S.; Bittman, R. J. Org. Chem., 1996, 61, 8709. 15. Leeuwenburgh, M.A.; Kulker, C.; Duynstee, H. I.; Overkleeft, H. S.; Van der Marel, G. A.; Van Boom, J. H. Tetrahedron, 1999, 55, 8253. 16. Kates, M. J.; Schauble, J. H.; J. Org. Chem., 1996, 61, 4164. 17. Han, X.; Crane, S. N.; Corey, E. J. Org. Lett., 2000, 2, 3437. 18. Kumar, P.; Naidu, S. V.; Gupta, P. J. Org. Chem., 2005, 70, 2843. 19. Ivanova, D. I.; Eremin, S. V.; Shvets, V. I. Tetrahedron, 1996, 52, 9581. 20. Wipf, P.; Graham, T. H. J. Am. Chem. Soc., 2004, 126, 15346. 21. Akira Tai, F.; Matsumura, H. C. C. J. Org. Chem., 1969, 34, 2180. 22. Campos, K. R.; Cai, D.; Journet, M.; Kowal, J. J.; Larsen, R. D.; Reider, P. J. J. Org. Chem., 2001, 66, 3634. 23.参考书:现代有机合成试剂<性质、制备和反应>;胡跃飞 付华 编著;化学工业出版社;ISBN 7-5025-8542-7
备注
53092-86-7
表征图谱

暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号