
结构式
| 物竞编号 | 0A8Y |
|---|---|
| 分子式 | (C25H23FNO4)2.Ca |
| 分子量 | 880.98 |
| 标签 | Livalo, 抑制剂 |
编号系统
CAS号:147526-32-7
MDL号:暂无
EINECS号:暂无
RTECS号:暂无
BRN号:暂无
PubChem号:暂无
物性数据
性状:白色固体。
毒理学数据
暂无
生态学数据
暂无
分子结构数据
暂无
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:4
3.氢键受体数量:12
4.可旋转化学键数量:14
5.互变异构体数量:无
6.拓扑分子极性表面积187
7.重原子数量:63
8.表面电荷:0
9.复杂度:626
10.同位素原子数量:0
11.确定原子立构中心数量:4
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:2
14.不确定化学键立构中心数量:0
15.共价键单元数量:3
性质与稳定性
暂无
贮存方法
暂无
合成方法
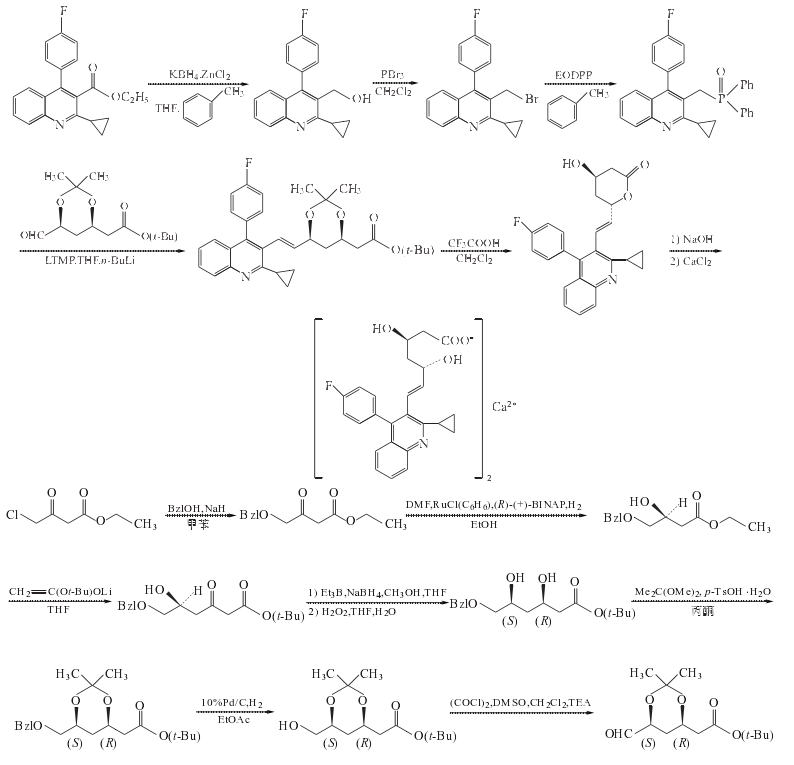
1. 2-环丙基-4-(4-氟苯基)喹啉-3-甲醇的制备
在反应瓶中加入THF196ml、ZnCl2 31.5g(0.23mol)和KBH4 24.5g(0.45mol),室温搅拌2h.加入2-环丙基-4-(4-氟苯基)喹啉-3-羧酸乙酯49g(0.15mol)的甲苯392ml溶液,缓慢升温至回流,搅拌回流反应20h.冷至室温,过滤,滤饼用甲苯加热回流洗涤,抽滤,合并有机层,加水后静置分层,水层用甲苯提取,合并有机层,用0.1mol/LNaOH水溶液洗涤,饱和盐水洗至中性.减压浓缩至干,剩余物用甲苯重结晶,得2-环丙基-4-(4-氟苯基)喹啉-3-甲醇27.7g,收率65%,mp128~130ºC(文献[4]收率70%,mp136~137ºC)
2. 3-溴甲基-2-环丙基-4-(4-氟苯基)喹啉的制备
在反应瓶中加入上步制备的化合物2-环丙基-4-(4-氟苯基)喹啉-3-甲醇25g(85mmol)和二氯甲烷200ml,降温至0ºC,在0ºC搅拌滴加三溴化磷(PBr3)16ml(170mmol)的二氯甲烷50ml溶液,温搅拌反应3h.蒸去二氯甲烷,余物缓慢倒入饱和NaHCO3水溶液中,至pH8~9,乙酸乙酯提取,机相用水洗至中性,压浓缩至干,余物用无水乙醇重结晶,3-溴甲基-2-环丙基-4-(4-氟苯基)喹啉27g,收率90%mp129~131ºC(文献[6]收率89%,mp140 ºC)
3. 2-环丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉的制备
在茄形瓶中加入上步制备的化合物3-溴甲基-2-环丙基-4-(4-氟苯基)喹啉27g(75.8mmol)、乙氧基二苯基膦35ml(150mmol)和甲苯270ml,回流12h.减压蒸干,剩余物用二氯甲烷/石油醚重结晶,得2-环丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉36.0g,收率定量,mp172~174ºC.
4. (3R,5S)-6-氧代-3,5-二羟基-3,5-O-异亚丙基己酸叔丁酯的制备
(1) 苄氧基乙酰乙酸乙酯的制备
在反应瓶中加入甲苯1000ml和55%~60%NaH73.5g(1.68~1.84mol),搅拌悬浮,降温至20~25ºC,往悬浮液滴加苯甲醇162g(155ml,1.498mol),在50min内滴加完,滴加过程中应用冰水浴微微冷却以保持温度在20~25ºC范围内,滴毕,将稠厚的浆状液于24ºC继续搅拌2h,氢气逸出终止.再在20~25ºC搅拌滴加4-氯乙酰乙酸乙酯125g,(103ml,1.7595mol),30min内滴加完,滴加过程中因放热反应,要适度冷却,以保持温度恒定在20~25ºC之间.滴毕,继续在03M搅拌反应19h.反应过程中用GC进行监测(GCl,tR:苯甲醇1.2min,化合物4-氯乙酰乙酸乙酯2.0min,化合物苄氧基乙酰乙酸乙酯7.1min)反应毕,于20~25ºC并微冷却下,在30min内加入2mol/L柠檬酸水溶液755ml(1.51mol),当该溶液加入30%以后,反应液成为非常稠厚浆状物,继续加入后,则反应液黏度降低.最后反应混合物pH为4.静置分层,水溶液层用甲苯100ml提取,合并有机层,用无水Na2SO4干燥,过滤,滤液减压蒸除溶剂,剩余油状物加庚烷(25ml×2)一起搅拌,分取庚烷层,仍为油状的庚烷层真空下浓缩,得粗品225g,将该粗品用双级薄膜蒸发器进行真空蒸馏[%控制第一蒸馏柱温140ºC,第二蒸馏柱温180ºC,真空度为66.66Pa(0.5Torr),得苄氧基乙酰乙酸乙酯纯品150g,收率84%,为无色黏滞油状物#纯度>99%
(2)(S)4-苄氧基-3-羟基丁酸乙酯的制备
在反应瓶中加入新鲜蒸馏的DMF900ml,在氩气通过液面以下保护下再加入(R)-(+)-2,2’-双(二苯膦基)-1,1’-联萘和苯基氯化钌二聚物7.0g(14.0mmol,28.0mmol RU),在氩气连续通过混合物的条件下,搅拌15min.然后反应瓶深深浸入预先加热到100ºC的油浴中,则溶液澄清,并变成红棕色清液.将其在氩气保护下冷至25ºC备用.此时,在30L不锈钢高压反应釜中加入无水乙醇10L和上步反应制备的化合物苄氧基乙酰乙酸乙酯保持釜内微带正压的氩气保护下加入上述制备的催化剂,加毕,密闭高压釜,用H2置换釜中氩气3次.然后充H2,搅拌加热至内温100ºC此时再通入H2调釜内H2压力为0.4MPa,恒压0.4MPa H2压力和100ºC下,一般反应12H则达反应终点.反应毕,降至室温,通过取样阀取小样进行分析,GC3分析表明>99%已氢化.HPLC分析粗品的光学纯度为97.4%.用N2将釜内气体清除3次,反应液放至蒸馏釜,高压釜用乙醇1L进行清洗,洗液与反应液合并,真空蒸除乙醇,高真空60ºC下蒸除DMF.剩余物用薄膜蒸发器于140~150ºC蒸馏柱温/13.33Pa条件下蒸出目的物(S)4-苄氧基-3-羟基丁酸乙酯,得(S)4-苄氧基-3-羟基丁酸乙酯9.70kg,收率96%,为无色油状物#如果在0~10ºC则固化结晶.
(3)(S)-6-苄氧基-5-羟基-3-氧代己酸叔丁酯的制备
在反应釜中加入二异丙胺9.4L,(71.7mol)和THF13.4L,搅拌下于0~10ºC1h内加入15%丁基锂的己烷溶液43.6L(69.8mol),该混合溶液在10ºC搅拌30min,再于-40ºC 40min内滴加乙酸叔丁酯9.0ml(66.8mol),再于-40ºC 30min内滴加上步制备的化合物(S)-6-苄氧基-5-羟基-3-氧代己酸叔丁酯4.0kg(16.8mol)的THF4L溶液,将混合物于-40ºC搅拌反应1h.用TLC跟踪确认反应达终点后,往反应混合物中加水(没有冷却)7.4L,30min加完,加完水后使反应混合物达-10ºC分出有机层,水层用甲苯(16L×2)提取,合并有机层,用无水Na2SO4干燥,过滤,滤液减压蒸除溶剂[,得半固体粗品(S)-6-苄氧基-5-羟基-3-氧代己酸叔丁酯7.3~7.7kg,内含有相当量的溶剂.
将粗品(S)-6-苄氧基-5-羟基-3-氧代己酸叔丁酯的半固体用异丙醚洗涤研磨捣碎后过滤,高真空下干燥,可得纯的分析试样,mp136~137ºC.
(4)3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯的制备
在反应釜中加入THF7.5L和上步制备的化合物(S)-6-苄氧基-5-羟基-3-氧代己酸叔丁酯7.54kg(16.8mol),搅拌溶解,20~25ºC15min内加入1mol/L三乙基硼的己烷溶液40.0L(40.0mol),反应吸热.加毕,将溶液搅拌10min.然后冷却至-60ºC,立刻加入硼氢化钠(NaBH4)1.9kg(50.4mol)(反应不放热),再在-60ºC 2.5h内滴加甲醇35L(强放热),滴加毕,在同温度下搅拌1h.TLC跟踪[展开剂为庚烷/乙酸乙酯(2:1)]确认反应终点,反应毕,15min内加水17L,将反应混合物转至装有搅拌器的200L分离器中,加入盐水17L,分相,水相用甲苯(33L×2)提取,分相后沉淀盐被沉积在分离器底部.合并有机相,用水22L洗涤#然后加入无水Na2SO4过夜.过滤掉干燥剂Na2SO4后,将滤液真空浓缩.剩余物加THF 34L溶解,在0~10 ºC和有效冷却下加入35%H2O2 7.5L,在1h内加完.将混合物继续搅拌30min,TLC跟踪,确认中间体硼酯反应完全,并形成了化合物3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯后,加入水16L和甲苯16L.再将混合物搅拌5min,静置分相.在有效冷却下,往分出的有机相加入NaHCO3 2.0kg,破坏过量的H2O2.混合物再搅拌10min.有机相用饱和的NaHCO3水溶液10L洗涤,然后用盐水5L洗涤,无水Na2SO4干燥,过滤,滤液减压蒸除溶剂,得油状剩余物7.0kg,与庚烷12L混合后转至装有搅拌器的100L分离器中,将混合物强烈搅拌1h.然后让其沉降2~3h.从产物相分取庚烷相,庚烷相再加入庚烷24L稀释,于5 ºC搅拌过夜.分取油状产物相,含在合并产物相的残留溶剂减压蒸除,得纯的3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯4.2kg(13.5mol),以(S)4-苄氧基-3-羟基丁酸乙酯计收率80%,产物3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯为特别淡的黄色油状物.
(5)(3R,5S)-6-苄氧基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯的制备
在反应釜中加入上步制备的化合物3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯11.1kg(35.76mol)和丙酮112L,开搅拌,冷至20~25ºC,加入2,2-二甲氧基丙烷7.35kg(8.68L,69.2mol)和对甲苯磺酸一水合物0.68kg(3.6mol),将起初的无色溶液于24ºC搅拌4h.在搅拌1~2h后溶液慢慢转变成黄色,用HLC跟踪,确认化合物3(R),5(S)-6-苄氧基-3,5-二羟基己酸叔丁酯已反应生成产物(3R,5S)-6-苄氧基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯后,加入固体NaHCO3 2.4kg(28.6mol),将悬浮液搅拌30min.然后将混合物过滤,滤液真空蒸除溶剂,得无色油状物11.5kg,粗品收率92%.
往200cm×30cm(外径)(体积141L)MPLC柱装入硅胶(70~210um)110kg和环己烷/乙酸乙酯(8:1)200L配成的浆料(溢流装柱).将上述粗品11.5kg溶于环己烷5L中,经过滤除去甲苯磺酸钠后的滤液进入上述色谱柱分离纯化(详细操作见文献[9]),经标准后处理和高真空干燥后,得纯的化合物(3R,5S)-6-苄氧基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯6.9kg,收率69%.纯度>99%(HPLC),产物为无色油状物.
(6) (3R,5S)-6-羟基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯的制备
在40L的搪瓷高压釜中加入上步制备的化合物3.25kg(9.27mol)和乙酸乙酯30L,用N2置换溶液中和釜中空气,再加入Pd/C(10%)催化剂0.32kg,用H2置换釜中N2后充H2(压力1MPa),搅拌下和恒氢气压力(1MPa)下于31~33氢化反应5h,经取样阀取试样进行TLC分析[展开剂:庚烷/乙酸乙酯(1:1)](3R,5S)-6-苄氧基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯斑点消失,产物(3R,5S)-6-羟基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯斑点明显,则反应达终点.用N2清除釜内氢气,过滤,滤液真空浓缩蒸除溶剂,剩余物加甲苯(1L×2),每次都真空蒸除甲苯,剩余物在高真空下干燥,得黏滞无色油状物(3R,5S)-6-羟基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯2.42kg,收率100%,纯度>98%.
(7) (3R,5S)-6-氧代-3,5-二羟基-3,5-O-异亚丙基己酸叔丁酯的制备
在干燥的四口反应瓶中加入干燥的二氯甲烷25ml和草酰氯1.0ml(11mmol),降温至-50~-60ºC,搅拌下慢慢往草酰氯的二氯甲烷溶液滴加DMSO1.7ml(1.72g,22mmol)与二氯甲烷5ml混合液,反应混合物搅拌2min,然后在同温度下5min内滴加上步制备的化合物(3R,5S)-6-羟基-3,5-O-异亚丙基-3,5-二羟基己酸叔丁酯2.6g(10mmol)与二氯甲烷10ml的混合液,加毕,连续搅拌15min.再加入三乙胺,将混合物搅拌5min.然后升至室温#加水50ml,搅拌片刻后静置分层,水层用二氯甲烷50ml反提取.合并有机层,用饱和Na2CO3水溶液洗涤,无水MgSO4干燥,过滤,滤液用旋转蒸发器浓缩至25ml,依次用1%稀盐酸、水、5%稀Na2CO3水溶液和水洗涤,然后将有机相浓缩至无溶剂,则得(3R,5S)-6-氧代-3,5-二羟基-3,5-O-异亚丙基己酸叔丁酯的粗品2.50g,收率97%,可直接用于下步反应.
5. (3R,5S,6E)-7-[2-环丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羟基-3,5-O-异亚丙基-6-庚烯酸叔丁酯的制备
在反应瓶中加入2,2,6,6-四甲基哌啶锂20ml(120mmol)和THF200ml,0ºC和N2保护下,搅拌滴加2.5mol/L正丁基锂,滴毕,于0ºC搅拌15min.冷却至-78ºC,滴加化合物(3R,5S)-6-氧代-3,5-二羟基-3,5-O-异亚丙基己酸叔丁酯50g(105mmol)的THF800ml溶液,搅拌1h.加入上步制备的中间体(3R,5S)-6-氧代-3,5-二羟基-3,5-O-异亚丙基己酸叔丁酯33.5g(130mmol)的THF200ml溶液,搅拌1h后升至室温反应过夜.加入饱和NaHCO3水溶液500ml终止反应,用乙酸乙酯提取,提取相用饱和盐水洗至中性,无水Na2SO4干燥,过滤,滤液减压蒸干,剩余物经硅胶柱色谱分离,得油状物37.5g,收率69%.
6. (4R,6S,E)-6-[2-环丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羟基-3,4,5,6-四氢-2H-吡喃-2-酮的制备
在反应瓶中加入上步制备的化合物(3R,5S,6E)-7-[2-环丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羟基-3,5-O-异亚丙基-6-庚烯酸叔丁酯2.52g(4.9mol)的二氯甲烷20ml溶液,搅拌降温至0ºC,滴加三氟乙酸5.6ml(73.5mmol)的二氯甲烷8ml溶液,滴毕,室温搅拌反应2h.加入饱和NaHCO3水溶液终止反应.调至pH7~8,用二氯甲烷提取,提取相用水洗涤,无水Na2SO4干燥,过滤,滤液减压浓缩至干,得1.97g,收率99.5%,mp128~130.
7. (3R,5S,6E)-(+)-7-[2-环丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羟基-6-庚烯酸钙盐(2:1)(匹伐他汀钙)的合成
在反应瓶中加入上步制备的化合物(4R,6S,E)-6-[2-环丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羟基-3,4,5,6-四氢-2H-吡喃-2-酮1.2g(3.0mmol)和无水乙醇20ml,搅拌溶解,再加入0.1mol/LNaOH水溶液32ml,室温搅拌反应1h.加入0.1mol/L盐酸调至pH7,减压蒸除溶剂,剩余物中加水20ml和无水氯化钙0.32g(2.9mmol),搅拌15min,后抽滤,滤饼用水洗涤,抽干,烘干,用异丙醇/水重结晶,得白色固体(3R,5S,6E)-(+)-7-[2-环丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羟基-6-庚烯酸钙盐(2:1)(匹伐他汀钙)1.0g,收率76.3%.
用途
本品为3-羟基-3-甲基戊二酰辅酶A还原酶抑制剂,可以减少肝脏合成胆固醇,使血浆低密度脂蛋白胆固醇的清除增加,可阻止动脉粥样硬化的进展.口服本品生物利用度高,半衰期长.具有肝脏选择性,与通过细胞色素P450系统代谢的药物发生相互作用的倾向更低.其降低甘油三酯的作用强于西立伐他汀、普伐他汀、氟伐他汀和辛伐他汀.临床上本品用于治疗高脂血症.
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
【1】 四川美康医药软件研究开发有限公司编著.药物临床信息参考.成都:四川出版集团四川科学技术出版社,2006:532. 【2】 Aoki T,et al.Arzneim-Forsch Drug Res,1997,47:904-909. 【3】 Miyachi N,et al.Tetrahedrom Lett,1993,34:8267-8270. 【4】 EP,304063.1989. 【5】 Wess G,et al.Tetrahedrom Lett,1990,31:2545-2548. 【6】 日本公开特许93-10700.(CA,1994,120:244704.) 【7】 EP,409281.1991. 【8】 蔡正艳等.中国医药工业杂志,2007,38:177-180. 【9】 Beck G,et al.Synthesis,1995,(8):1014-1018. 【10】 Manusco A J,et al.J Org Chem,1978,43:2480-2482. 【11】 Muller P,et al.Terachedrom Lett,1981,22:2361. 【12】 周伟澄主编.高等药物化学选论.北京:化学工业出版社,2006:314-316. 【13】 Hiyama T,et al.Bull Chem Soc Jpn,1995,68:364-444.
备注
暂无
表征图谱
暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号