
结构式
| 物竞编号 | 0ECF |
|---|---|
| 分子式 | C15H22FN3O6 |
| 分子量 | 359.35 |
| 标签 | 5-脱氧-5-氟-N-[(戊氧基)羰基]-胞嘧啶核苷, 5-脱氧-5-氟-N-[(戊氧基)羰基]-胞嘧啶核甙, 希罗达, Pentyl [1-(3,4-dihydroxy-5-methyl-oxolan-2-yl)-5-fluoro-2-oxo-pyrimidin-4-yl]aminoformate, Xeloda, 药物 |
编号系统
CAS号:154361-50-9
MDL号:暂无
EINECS号:暂无
RTECS号:暂无
BRN号:暂无
PubChem号:暂无
物性数据
1.性状:白色固体。
2.熔点(ºC):110~121
毒理学数据
暂无
生态学数据
暂无
分子结构数据
暂无
计算化学数据
1.疏水参数计算参考值(XlogP):0.6
2.氢键供体数量:3
3.氢键受体数量:7
4.可旋转化学键数量:7
5.互变异构体数量:4
6.拓扑分子极性表面积121
7.重原子数量:25
8.表面电荷:0
9.复杂度:582
10.同位素原子数量:0
11.确定原子立构中心数量:4
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
暂无
贮存方法
暂无
合成方法
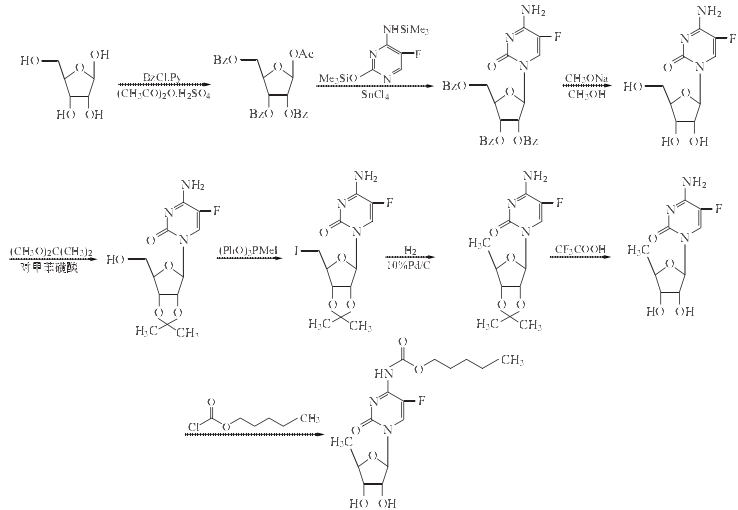
1. 1-O-乙酰-2,3,5-三-O-苯甲酰-β-D-核糖的制备
在反应瓶中加入无水甲醇220ml,再将D-核糖10g(67mmol)和3ml(0.3g/ml)无水氯化氢的甲醇溶液加入,室温搅拌90min.加入20ml吡啶,控温在50ºC以下浓缩至干,再加入20ml吡啶同样方法蒸干.蒸干的剩余物溶于无水二氯甲烷54ml和吡啶120ml,在冰浴冷却下(0ºC)搅拌下,慢慢滴加苯甲酰氯40ml(0.34mol).滴毕于冰箱放置48h(4ºC).倒入水266ºC中,分出有机层,水层用氯仿(54ml×3)提取,合并有机层,用饱和NaHCO3溶液(54ml×2)洗涤,洗液用40ml氯仿逆提取.合并有机层,用无水MgSO4干燥过夜,过滤,滤液减压蒸干(微量的吡啶用甲苯10ml×2共沸除去).于冰浴冷却下向剩余物加入乙酸16ml、乙酸酐38ml,然后搅拌下滴加浓硫酸5.4ml.滴毕移开冰浴,室温搅拌2h.于冰箱放置(4ºC)过夜.向反应混合物中加入200ml冰水#析出固体,过滤,水洗,干燥,用乙醇/乙酸乙酯(体积比5:2)40ml重结晶,得白色结晶1-O-乙酰-2,3,5-三-O-苯甲酰-β-D-核糖24g,收率71%,mp128~130ºC(文献[7]报道mp131~132ºC,收率70%).
2. 2’,3’,5’-O-三苯甲酰-5-氟胞苷的制备
在反应瓶中加入5-氟胞嘧啶5.85g(4.5mmol)和六甲基二硅胺烷30ml,搅拌悬浮,再加入三甲基氯硅烷,将混合物搅拌回流2h.于90ºC以下减压浓缩回收大部分的六甲基二硅胺烷,最后在1.6×103Pa的压力下完全蒸干.冷至室温,加入20g(40mmol)和无水1,2-二氯乙烷350ml,于0ºC搅拌下滴加无水四氯化锡[4.7ml(41mmol)的无水1,2-二氯乙烷(35ml)溶液,滴毕,室温继续搅拌过夜.在搅拌下慢慢加水20ml,再用约70ml饱和NaHCO3溶液调至pH8.抽滤,滤饼用1,2-二氯乙烷(15ml×4)洗涤,抽干,合并二氯乙烷溶液,无水MgSO4干燥,过滤,滤液浓缩至干.剩余物用乙醇200ml重结晶,得白色结晶2’,3’,5’-O-三苯甲酰-5-氟胞19.5g,收率86%,mp188~190ºC(文献[13] 报道mp191~192ºC,收率80%).
3. 5-氟胞苷的制备
在反应瓶中加入2’,3’,5’-O-三苯甲酰-5-氟胞苷30g(52mmol)和甲醇400ml,搅拌溶解,搅拌下滴加0.5mol/L甲醇钠的甲醇溶液约320ml,室温搅拌24h.用1.4mol/L氯化氢甲醇溶液中和至pH6.5左右,过滤除去NaCl,减压浓缩至干.剩余物用甲醇20ml溶解,过滤,滤液蒸干.剩余物溶于水70ml中.用氯仿(10ml×4)提取苯甲酸,水层蒸发至干,得白色固体5-氟胞苷13g,收率95%,无需纯化,直接用于下步反应.
4. 2’,3’-O-异亚丙基-5-氟胞苷的制备
在反应瓶中加入丙酮225ml和5-氟胞苷13g(50mmol),搅拌悬浮,加入对甲苯磺酸12g(63mmol)和2,2-二甲氧基丙烷30ml,室温搅拌2h.加入碳酸氢钠5.3g使反应液呈碱性.过滤,固体用丙酮(5ml×2)洗,滤液和洗液合并,浓缩至干,用乙酸乙酯100ml重结晶,得白色固体2’,3’-O-异亚丙基-5-氟胞苷13g,收率90%,mp184~186ºC,(文献[8]报道mp182~184ºC,收率92%)
5. 5’-碘代-2’,3’-O-异亚丙基-5-氟胞苷的制备
在反应瓶中加入2’,3’-O-异亚丙基-5-氟胞苷32g(106mmol)、三苯基亚磷酸酯甲基碘化物60g(133mmol),搅拌溶解,室温搅拌反应1.5h.加入100ml甲醇,继续搅拌30min,将溶液浓缩至干.剩余物呈油状,将其溶于乙酸乙酯700ml中,依次用0.3mol/L硫代硫酸钠溶液(700ml×2)、水(700ml×2)洗涤,无水MgSO4干燥,过滤,滤液浓缩至干.剩余物溶于400ml热乙酸乙酯中,加入正己烷1200ml,析出结晶0ºC下放置过夜,过滤,得白色结晶30g, 收率70%,mp192~193 ºC.
6. 5’-脱氧-2’,3’-O-异亚丙基-5-氟胞苷的制备
在反应瓶中加入4.8g(11.65mmol)和甲醇50ml,搅拌溶解,加入三乙胺2ml和10%(质量分数)Pd/C2g,于常压通H2 1h左右,经TLC检测原料斑点消失加H2反应到终点.过滤,滤液浓缩至干,剩余物加入乙酸乙酯20ml溶解. 放置过夜,滤除固体杂质,滤液浓缩至10ml左右.再放置过夜,滤除固体杂质,滤液浓缩至干,真空干燥,得白色泡沫状固体5’-脱氧-2’,3’-O-异亚丙基-5-氟胞苷3g,收率90%,无熔点.
7. 5’-脱氧-5-氟胞苷的制备
在反应瓶中加入3.1g(10.9mmol)和三氟乙酸/水(9:1)混合液20ml,搅拌40min.蒸干溶液,剩余物中残留的水及三氟乙酸用乙醇(5ml×2)共沸蒸除.剩余物中加入乙酸乙酯40ml溶解,再加入三乙胺2ml使溶液呈碱性,析出晶体.放置过夜.过滤,滤饼用乙酸乙酯5ml洗,真空干燥,得白色结晶1.9g,收率71%,mp192~193 ºC.
8. 5’-脱氧-5-氟-N4-(戊氧羰酰基)胞苷(卡培他滨)的合成
在反应瓶中加入5’-脱氧-5-氟胞苷3.0g(12.2mmol)和DMF90ml,搅拌溶解,冰浴(0ºC) 冷却下搅拌滴加正戊
基氯甲酸酯2ml(12.8mmol),滴毕移去冰浴,用650w的微波加热30~50s.TLC检测原料斑点消失.将反应液浓缩至干,加入氯仿100ml和1.7mol/LNaCl溶液50ml,分出有机层,用无水NaSO4干燥过夜,过滤,滤液浓缩至干.剩余物用乙酸乙酯40ml重结晶,得白色固体5’-脱氧-5-氟-N4-(戊氧羰酰基)胞苷(卡培他滨)4g,收率90%,反应
总收率22%,mp110~115ºC,(文献[2,3]报道mp110~121 ºC,收率55%,总收率12%)
用途
本品是5-氟尿嘧啶(5-FU)的前药,在体内转化成5-FU),从而发挥抗癌作用.抗癌活性与5'-脱氧-5-氟尿嘧啶(5'-DFUR)相似,且活性与使用总剂量有关,而与治疗使用时间长短无关.床用于直肠癌和结肠癌的一线治疗,同时也可作为转移型乳腺癌的单独或联合治疗.品无细胞毒性,疗效好,安全.
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
【1】 Merck Index.13th:1759. 【2】 US,5472949.1995. 【3】 EP,602454.1994. 【4】 Matsuda A,et al.Synthesis,1981,12:748. 【5】 董辉等.中国医药工业杂志,2002,33:108-109. 【6】 Nahar P.Tetrahedre Lett,1974,38:7253-7254. 【7】 Niedballa U,et al.J Org Chem,1974,39:3654-3660. 【8】 US,4071680.1978. 【9】 Verheyden J P H,et al.J Org Chem,1970,35:2319-2346. 【10】 尤启冬,林国强主编.手性药物---研究与应用.北京:化学工业出版社,2004:551-553. 【11】 余建鑫等.中国药物化学杂志,2005,15:173~175,187.
备注
暂无
表征图谱
暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号