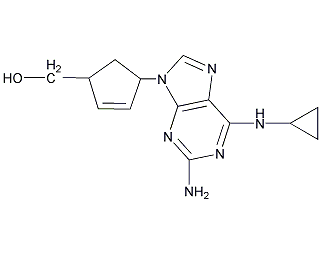
结构式
| 物竞编号 | 005B |
|---|---|
| 分子式 | C14H18N6O |
| 分子量 | 286.33 |
| 标签 | 赛进, 硫酸阿巴卡韦, AbacavirSulfate, 抗病毒药 |
编号系统
CAS号:136470-78-5
MDL号:暂无
EINECS号:暂无
RTECS号:暂无
BRN号:暂无
PubChem号:暂无
物性数据
性状:白色固体泡沫
毒理学数据
暂无
生态学数据
暂无
分子结构数据
1、 摩尔折射率:75.80
2、 摩尔体积(cm3/mol):167.6
3、 等张比容(90.2K):501.7
4、 表面张力(dyne/cm):80.1
5、 极化率(10-24cm3):30.05
计算化学数据
1.疏水参数计算参考值(XlogP):0.9
2.氢键供体数量:3
3.氢键受体数量:6
4.可旋转化学键数量:4
5.互变异构体数量:6
6.拓扑分子极性表面积102
7.重原子数量:21
8.表面电荷:0
9.复杂度:414
10.同位素原子数量:0
11.确定原子立构中心数量:2
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
暂无
贮存方法
暂无
合成方法
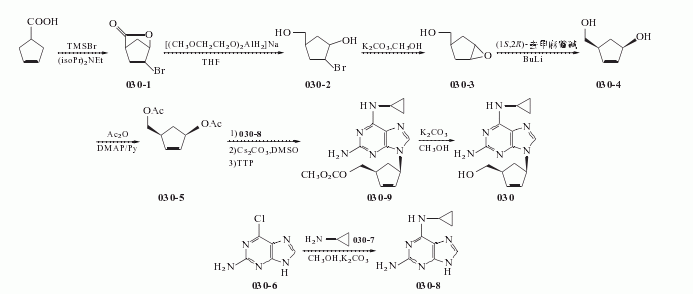
1.乙酰氨基环己-2-烯甲醇乙酸酯和氢氧化钡八水合物溶于水,在氮气保护下搅拌反应,用二氧化碳中和反应液,过滤,滤液浓缩。得到的物质和2-氨基4,6-二氯嘧啶及三乙胺在正丁醇中搅拌反应,加入盐酸,浓缩至干,得到的物质溶于氯仿。过滤除去未反应的2-氨基-4,6-二氯嘧啶,用氯仿洗涤。氯仿层合并,浓缩,经过柱层析,将柱层析分离得到的产物和乙酸钠三水合物溶于乙酸水溶液中,缓慢加入由4-氯苯重氮盐酸盐、浓盐酸、水和硝酸钠所形成的冷的溶液,搅拌,过滤收集沉淀,水洗,得到产物4-[2-氨基-6-氯-5-(4,5-(4-氯苯基)偶氮-4-嘧啶基]氨基-2-环己烯-1-甲醇。将该产物悬浮于乙醇中,加入乙酸水溶液,在氮气下回流,分批加入锌粉反应。过滤除去未反应的锌,滤液浓缩,得到的物质柱层析提纯,得4-[(2,5-二氨基-4-氯-6-嘧啶基)氨基]-2-环己烯-1-甲醇。将其溶于乙酸二乙氧基甲酯中,回流。在真空下蒸出溶剂,加入二氧六环和盐酸,室温搅拌、冷却,用氢氧化钠中和至pH=7,再用3∶1的氯仿/甲醇提取。有机层用硫酸镁干燥,过滤,浓缩,得到的物质柱层析,得4-(2-氨基-6-氯-9-N-嘌呤-9-基)-2-环己烯-1-甲醇。该产物溶于乙醇,加入环丙胺,在氮气下反应6h。浓缩,加入氯仿和饱和碳酸氢钠溶液。水层用氯仿提取,浓缩,柱层析分离,即得产物。
2.
推荐文献[8,10,25]的合成路线和方法。
(1)exo-(+-)-6-溴-氧杂双环[2,2,1]庚烷-3-酮(030-1)的制备
在反应瓶中加入二甲基亚砜(DMSO)0.64ml(9.0mmol)和三氯甲烷(CHCL3)15ml,搅拌下降温至0 ºC ,搅拌滴加溴代三甲基硅烷(trimethysily bro-mide,TMSBr)1.2ml(9.1mmol),保持在0 ºC 搅拌3h.再滴加环戊-6-烯羧酸(cyclopent-3-ene-carboxylicacid)0.784g(7mmol)的三氯甲烷5ml溶液,于15min内滴完,搅拌反应10min.向反应混合物中加入二异丙基乙胺1.57ml(9.0mmol),然后搅拌回流反应12h.冷却至室温,应混合物用CH2CL230ml稀释,搅拌
均匀后,用水(40ml*2)洗涤,再用盐水40ml洗涤,振摇后静置分层,有机层用无水MgSO4干燥,过滤,将滤液减压蒸除溶剂,剩余物进行真空蒸馏,收集150~160 º/13.3Pa馏分,得油状物030-1 1.276g,收率95%;bp150~160 ºC /0.1mmHg(13.3Pa).
(2).(1a,3B,4a)-(+-)-3-溴-4-羟基环戊烷甲醇(030-2)的制备
在反应瓶中加入化合物030-1 1.74g(9.11mmol)和干燥的THF20ml,搅拌溶解,在氩气保护下,搅拌降温至-25 ºC (用CCl4/干冰浴冷却),保持在-25 ºC 下搅拌滴加3.46mol/dm3的双(2-甲氧基乙氧基)二氢化铝钠{[(CH3OCH2CH2O)2ALH2]Na}的甲苯溶液2.7ml(9.3mmol),控制15s内滴完,滴毕,继续搅拌2min.向反应混合物加水0.34ml,随即加入150g/L的NaOH水溶液0.34ml,再加水1ml,将反应混合物过滤,滤液用乙酸乙酯10ml稀释,再用饱和NH4Cl水溶液稀释,用乙酸乙酯(15ml*2)提取,合并有机层,无水MgSO4干燥,过滤,将滤液减压下蒸除溶剂,得剩余物030-2粗品1.67g,为无色油状物,收率94%,无需进一步纯化,直接用于下步反应.Rf0.25(20%乙酸乙酯/乙醚).
(3).(1a,3a,5a)-6-氧杂双环[3,1,0]己烷-3-甲醇(030-3)的制备
在反应瓶中加入上步粗制的化合物030-2 187mg和甲醇2ml,搅拌溶解,往该溶液中加入K2CO3 451mg(3.26mmol)(加时,一次性加入),将混合物搅拌反应12h.过滤,将滤液减压蒸除溶剂,剩余物经硅胶柱色谱纯化(洗脱剂:15%乙醚/石油醚),首先得油状的内消旋环氧化物030-3 41mg,收率35%,随后得未变化的溴代二醇(030-2)92mg.030-3:Rf0.30(乙醚).
(4)(1S)-顺式-4-羟基-2-环戊烯-1-甲醇双乙酸酯[即(1S,4R)-1-乙酰氧基-4-(乙酰氧甲基)-环戊基-2-烯](030-5)的制备
在反应瓶中加入(1S,2R)-去甲麻黄碱[(1S,2R)-norephedrine]的苯/THF溶液[888mg(5.87mmol)(1S,2R)-去甲麻黄碱溶于体积比为15:10的苯/THF],搅拌下于0 ºC 滴加丁基锂的己烷溶液(2.5mol/L)6.5ml(16.2mmol),滴毕,搅拌30min.滴加化合物030-3 200mg(1.75mmol)的THF 3ml溶液,在15min滴加完.滴毕,自然升温至室温,在室温搅拌反应过夜.加入甲醇10ml,反应液搅拌均匀后,通过硅藻土[Celite 545(Fluka)]过滤,滤液在减压下蒸除溶剂,剩余物用1gSiO2吸附,然后用抽吸快速色谱纯化(度洗脱剂:(乙醚-10%乙醚/乙酸乙酯,
40ml fractions).得油状物030-4 115mg,收率57%.
(5).(1S,4R)-4-羟基-3-环戊烯-1-甲醇双乙酸酯[即(1S,4R)-1-乙酰氧基-4-(乙酰氧甲基)-环戊基-2-烯](030-5)的制备
在反应瓶中加入030-4 391mg(3.4mmol)、乙酸酐1.3ml(14mmol)和二甲基氨基吡啶(DMAP)40mg(0.3mmol)的吡啶20ml溶液,上述物料时应在搅拌下,并控制温度在25 ºC ,加好物料仍控制温度在25 ºC 搅拌反应12h.将反应混合物用乙醚100ml释,搅拌均匀后,反应混合物的乙醚溶液用水100ml洗涤,再用饱和的NH4CL水溶液(100ml*2)洗涤,最后用盐水100ml洗涤.分取有机层,用无水MgSO4干燥,过滤,滤液于减压下蒸除溶剂,剩余物经硅胶柱色谱纯化(洗脱剂:35%乙醚/石油醚混合溶剂),得白色半固体的030-5 660mg,收率97%,Rf0.30(35%乙醚/石油醚).
(6).2-氨基-6-(环丙氨基)9H-嘌呤(030-8)的制备
在反应瓶中加入2-氨基-6-氯嘌呤(030-6)(外购或按文献[21~23]的方法制备)5.0g(0.0295mol)和甲醇15ml,搅拌混合,再加入环丙胺(030-7)12.5ml(10.3g,0.1804mol),将该悬浮液加热至回流,搅拌回流反应28h.降温至40 ºC ,然后真空下蒸出过量的环丙胺和溶剂,得黄-橙色泡沫状物。将泡沫状物用异丙醇200ml和甲醇50ml溶解,搅拌下加入K2CO3 5.0g(0.0362mol),冷至0~5 ºC ,在该温度下搅拌30min.过滤,滤液浓缩,剩余物于45 ºC 真空下干燥64h.得橙色泡沫状物7.11g.将该泡物用乙腈71.1ml和异丙醇71.1ml重结晶,过滤,滤饼在真空下干燥(45 ºC)则得到淡橙色固体030-8 4.42g(或者可先用小样摸索重结晶,即取泡沫状物
1g比例用乙腈和异丙醇混合溶剂重结晶),收率:78.8%,mp150~152 ºC.
(7).(1S,4R)- 4-[2-氨基-6-(环丙氨基)-9H-嘌呤-9-基]环戊烯(基)-1-甲醇(阿巴卡韦)030的合成
在反应瓶中加入030-8 1.0g(0.0053mol)、碳酸铯1.75g(0.0054mol)和干燥的DMSO 50ml,在N2保护下,搅拌升温至60 ºC ,并在该温度下搅拌2h.将混合物冷至室温,然后加入四(三苯基磷)合钯(TTP)[0.85(0.00074mol)]和化合物030-5[0.79g(0.0034mol)]的DMSO(10ml)溶液,搅拌加热至65 ºC ,持65 ºC 下搅拌反应2.25h.混合液中即含化合物030-9.
向混合液中加入甲醇100ml和K2CO3 2.10g,将混合物于40 ºC 搅拌反应45min.此时有固体沉淀,将其通过硅藻土层过滤,滤液于90 ºC 下真空蒸发至小体积,剩余胶状物用二氯甲烷(100ml*2)捣磨,得棕色固体剩余物经硅胶(Merck 9385)柱色谱纯化[洗脱剂:二氯甲烷/甲醇(积比9:1)],得黄色泡沫状物030 0.26 g,收率26.8
用途
酶抑制剂,用于人免疫缺陷病毒。与其他核苷类逆转录酶抑制剂一样,代谢成为具活性的三磷酸酯,抑制人免疫缺陷病毒逆转录酶的作用。与其他抗艾滋病药物联合应用,治疗HIV感染的患者。
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
[1] Merck Index. 13th : 1. [2] Foster R H,et al.Drugs,1998,55:729-736 [3] Staszewski S,et al.J Am Med Assoc,2001,285:1155. [4] EP,349242.1990. [5]US,5034394.1991. [6]Crimmins M T,et al.J Org Chem,1996,61:4192-4193 [7]Huff J R,Bioorg Med Chem,1999,7:2667-2669 [8] Hodgsom D M,et al.J Chem Soc Perkin Trans I,1994,3373-3377. [9] WO,9115490.1991. [10]WO,9924431.1999. [11]Crimmins M T,et al.J Org Chem,2000,65:8499-8509. [12]Olivo H F,et al.J Chem Soc Perkin Trans I,1998,391-392. [13]日本公开特许2003-55377. [14]Crimmins M T,et al.Org Lett,2000,2:1065. [15]Jagt J C.J Org Chem,1974,39:564. [16]US,4268672.1981. [17]WO,9521161.1995. [18] EP,434450.1991. [19] 王挣等。中国医药工业杂志,2005,36:720-722. [20]周伟澄主编。高等药物化学选论。北京:化学工业出版社,2006.180-181. [21]WO,9315075.1993. [22] DE,4342131. [23] DE,4415196. [24] EP662970 [25] Hodgson D M,et al.J Chem Soc Perkin Trans,I,1993,1543-1544. [26] 尤启冬。林国强主编。手性药物—研究与应用。北京:化学工业出版社,2004:263-265. [27] 日本公开特许,96-092252
备注
暂无
表征图谱

暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号