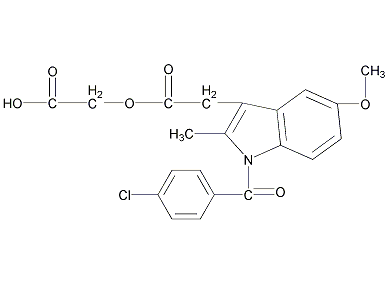
结构式
| 物竞编号 | 00MX |
|---|---|
| 分子式 | C21H18ClNO6 |
| 分子量 | 415.83 |
| 标签 | 1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1H-吲哚-3-乙酸羧甲酯, 消炎痛醋酯, Rantudil, BAY f4975, TVX-1322, Solart, 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid carboxymethyl ester, 非甾体消炎药, 药物, 中枢神经系统用药 |
编号系统
CAS号:53164-05-9
MDL号:MFCD00151473
EINECS号:258-403-4
RTECS号:NL3521400
BRN号:暂无
PubChem号:24890474
物性数据
1.性状:从石油醚得淡黄色细微结晶。
2.熔点:150-153℃。
毒理学数据
急性毒性LD50雄、雌小鼠,雄、雌大鼠(mg/kg):55.5,18.42,24.2,30.1口服;34.1,51.1,38.1,28.3静脉注射。
生态学数据
暂无
分子结构数据
1、 摩尔折射率:106.05
2、 摩尔体积(cm3/mol):305.4
3、 等张比容(90.2K):809.7
4、 表面张力(dyne/cm):49.4
5、 极化率(10-24cm3):42.04
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:1
3.氢键受体数量:6
4.可旋转化学键数量:7
5.互变异构体数量:无
6.拓扑分子极性表面积94.8
7.重原子数量:29
8.表面电荷:0
9.复杂度:620
10.同位素原子数量:0
11.确定原子立构中心数量:0
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
暂无
贮存方法
暂无
合成方法
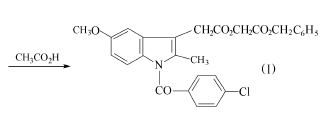
方法1:以在吲哚美辛的合成中得到的N-(4-甲氧基苯基)-4-氯苯甲酰肼为原料,和(3-乙酰基丙酰氧基)乙酸苄酯(其制备见方法3)环合,得到[1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1H-吲哚-3-乙酰氧基]乙酸苄酯(I)。得到的苄酯(I)溶于冰醋酸中,在室温下在钯-炭催化剂上进行加氢,当氢吸收停止后,过滤去催化剂,滤液减压浓缩,残液加入石油醚中重结晶,得阿西美辛精品,熔点149.5~150.5℃,收率93%。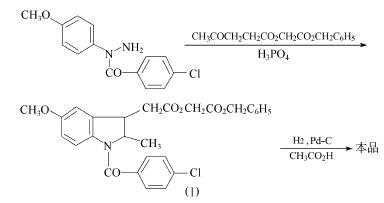 方法2:以吲哚美新为原料。
方法2:以吲哚美新为原料。
在搅拌下,将180g(0.503mo1)吲哚美新溶于900ml无水二甲基甲酰胺,加入35g(0.253mo1)磨细和干燥的碳酸钾,在50℃下搅拌45min。在15min中,滴入128g(0.56mo1)溴乙酸苄酯,在50℃下搅拌3h。过滤,滤液浓缩,剩余物溶于氯仿,用水洗至中性,无水硫酸钠干燥。浓缩,异丙醚-乙酸乙酯重结晶,得228g化合物(I),收率89.5%,熔点96℃。
60g(0.12mo1)化合物(I)溶于700ml乙酸乙酯,加入6g 5%钯-炭催化剂,在40℃下氢化1h,吸收约2.921L氢气(20℃)。过滤,浓缩,往剩余物加入石油醚(40~60℃),使结晶完全,并在冰箱中放置12h。过滤,先空气中干燥,后真空干燥,得46g阿西美辛,收率93%,熔点150~153℃。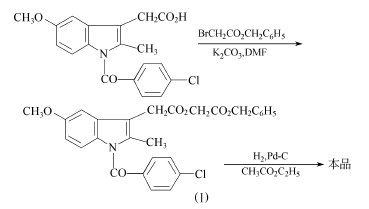
方法3:以对甲氧基苯肼和(3-乙酰基丙酰氧基)乙酸苄酯为原料。(3-乙酰基丙酰氧基)乙酸苄酯的制备。93.5g(0.68mo1)干燥碳酸钾悬浮于650ml无水二甲基甲酰胺,加热至40℃,在70min内滴入116.1g(1.0mo1)3-乙酰基丙酸。加毕,冷至30℃,加入229.1g(1.0mo1)2-溴乙酸苄酯。在50℃下反应4h。过滤除去无机盐,用甲苯提取。提取液浓缩,剩余物溶于400ml二氯甲烷,水洗至中性,无水硫酸钠干燥。浓缩,剩余物溶于。1000ml异丙醚,加入冷至-25℃的1500ml石油醚(40~60℃),于-15℃下放置1h。过滤,在22℃下真空干燥,得235g(3-乙酰基丙酰氧基)乙酸苄酯,收率89%,熔点31.5~32.5℃,沸点176~178℃/1.20kPa。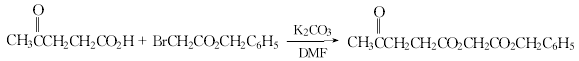
26.3g(0.19mo1)对甲氧基苯肼溶于125ml冰乙酸和450ml水,在搅拌下,滴入50g(0.19mo1)(3-乙酰基丙酰氧基)乙酸苄酯,再搅拌1h。用甲苯提取,提取液水洗至中性,干燥,浓缩,得75.5g棕色油状的化合物(Ⅱ),直接用于下列反应。
将化合物(Ⅱ)直接溶于125ml无水吡啶,加入500ml无水乙醚,再在0℃加入43.5g对氯苯甲酰氯在250ml无水乙醚的溶液,在氮气保护下回流2h,冷却,水洗至中性,干燥,浓缩,得95g化合物(Ⅲ)的粗品。无需提纯,直接用于下步反应。
将化合物(Ⅲ)直接溶于380ml乙酸,在80℃下加热5min。减压浓缩,剩余物溶于氯仿,用碳酸氢钠溶液洗,并水洗至中性。用氧化铝层析,氯仿洗脱。浓缩后用乙醚重结晶,得10.6g化合物(I)。从对甲氧基苯肼计,收率11%。化合物(I)再如方法2还原为阿西美辛。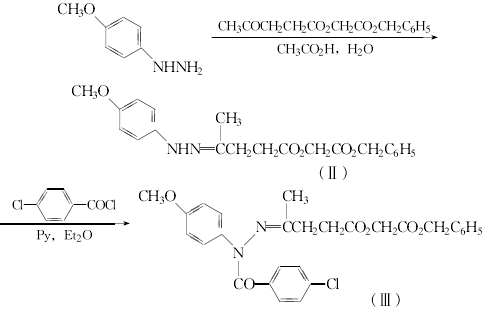
 方法4:对甲氧基苯肼和(3-乙酰基丙酰氧基)乙酸苄酯也可先环合,再对氯苯甲酰化。
方法4:对甲氧基苯肼和(3-乙酰基丙酰氧基)乙酸苄酯也可先环合,再对氯苯甲酰化。
45.7g(0.26mo1)对甲氧基苯肼盐酸盐和69.2g(0.26mo1)(3-乙酰基丙酰氧基)乙酸苄酯溶于260ml乙酸,先在28℃搅拌42h,再在50℃搅拌5h。蒸出150~180ml溶剂后,加入500ml冰水,用4×100ml甲苯提取。提取液合并,用碳酸氢钾溶液洗至无酸,无水硫酸钠干燥。过滤,浓缩,得92g粗品。经蒸馏,可得76g化合物(IV),收率80%,沸点165℃(≤100Pa)。
77.5g(0.44mo1)对氯苯甲酰氯溶于275ml专用石油(沸点180~220℃)中加热至沸,在20min中,滴加41g(0.1lmo1)化合物(Ⅳ)。加毕,在190-195℃加热2~3h,至无氯化氢放出。冷至40℃,加入200ml乙醚。在室温下结晶后,再放入冰箱过夜。过滤收集结晶,用异丙醚洗,再用乙酸乙酯和异丙醚重结晶,得43.3g化合物(I),收率78%。化合物(I)再如方法2还原为阿西美辛。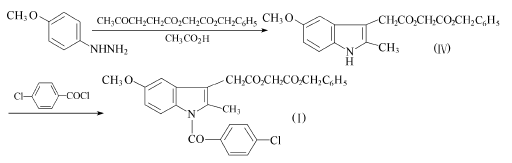
用途
非甾体消炎镇痛药,有明显抑制炎症的作用。适用于风湿性关节炎、类风湿性关节炎、肌纤维组织炎、坐骨神经痛等的治疗。
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
None
备注
None
表征图谱

暂无图谱
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号