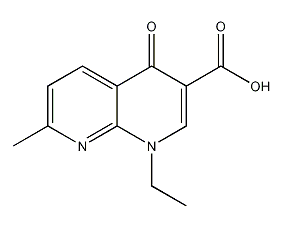
结构式
| 物竞编号 | 04LW |
|---|---|
| 分子式 | C12H12N2O3 |
| 分子量 | 232.24 |
| 标签 | 1,4二氢-1-乙基-7-甲基-1,8-萘-4-1-3-羧酸, 1-乙基-1,4-二氢-7-甲基-4-氧代-1,8-萘-3-羧酸, 1,4-Dihydro-1-ethyl-7-methyl-1,8-naphthyridin-4-one-3-carboxylic acid, 1-Ethyl-1,4-dihydro-7-methyl-4-oxo-1,8-naphthyridine-3-carboxylic acid |
编号系统
CAS号:389-08-2
MDL号:MFCD00006884
EINECS号:206-864-7
RTECS号:QN2885000
BRN号:750515
PubChem号:24897864
物性数据
一、物性数据
性状:几乎白色或极浅黄色粉末
密度(g/mL,25/4℃): 不可用
相对蒸汽密度(g/mL,空气=1):不可用
熔点(ºC):229-230
沸点(ºC,常压):不可用
沸点(ºC,5.2kPa): 不可用
折射率: 不可用
闪点(ºC): 不可用
比旋光度(º): 不可用
自燃点或引燃温度(ºC): 不可用
蒸气压(kPa,25ºC): 不可用
饱和蒸气压(kPa,60ºC): 不可用
燃烧热(KJ/mol):不可用
临界温度(ºC): 不可用
临界压力(KPa): 不可用
油水(辛醇/水)分配系数的对数值:不可用
爆炸上限(%,V/V):不可用
爆炸下限(%,V/V): 不可用
溶解性:溶于氯仿,微溶于醇、强碱液,几乎不溶于水和醚
毒理学数据
二、毒理学数据:
急性毒性:不可用 。
生态学数据
三、生态学数据:
1、其它有害作用:该物质对环境可能有危害,对水体应给予特别注意。
分子结构数据
1、 摩尔折射率:60.07
2、 摩尔体积(m3/mol):174.4
3、 等张比容(90.2K):484.1
4、 表面张力(dyne/cm):59.3
5、 极化率(10
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:1
3.氢键受体数量:5
4.可旋转化学键数量:2
5.互变异构体数量:5
6.拓扑分子极性表面积70.5
7.重原子数量:17
8.表面电荷:0
9.复杂度:378
10.同位素原子数量:0
11.确定原子立构中心数量:0
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
无气味。几乎无味。对光和空气敏感
贮存方法
密封保存。
合成方法
由2-甲基吡啶制得2-氨基-5-甲基吡啶,再与原甲酸乙酯及丙二酸二乙酯缩合成N-(2-甲基-5-氨基吡啶)甲叉丙二酸二乙酯,然后将其在260-270℃环合后再加入氢氧化钠稀溶液中水解,得到7-甲基-1,8-萘啶-4-羟基-3-羟酸。最后用溴乙烷进行N-烷基化并异构成萘啶酮酸。
用途
该品为抗菌剂,能抑制细菌DNA、RNA的合成,用于治疗革兰氏阴性细菌所致的尿道感染。萘啶酮酸是喹诺酮类的第一代抗菌药代表,第二代则以吡哌为代表。第三代喹诺酮类抗菌药物的代表是氟哌酸。
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
暂无
备注
暂无
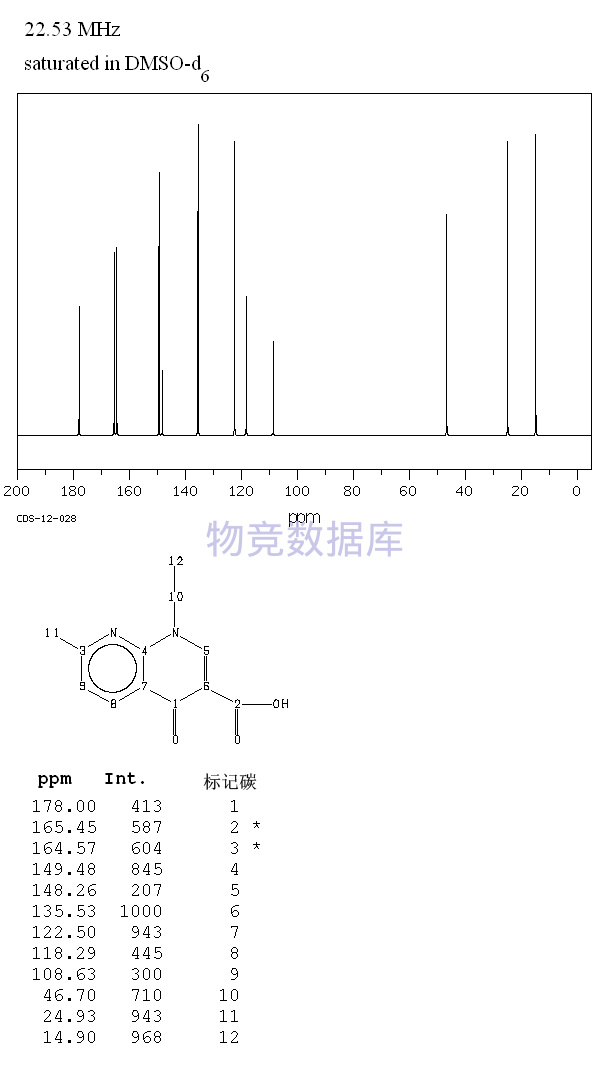
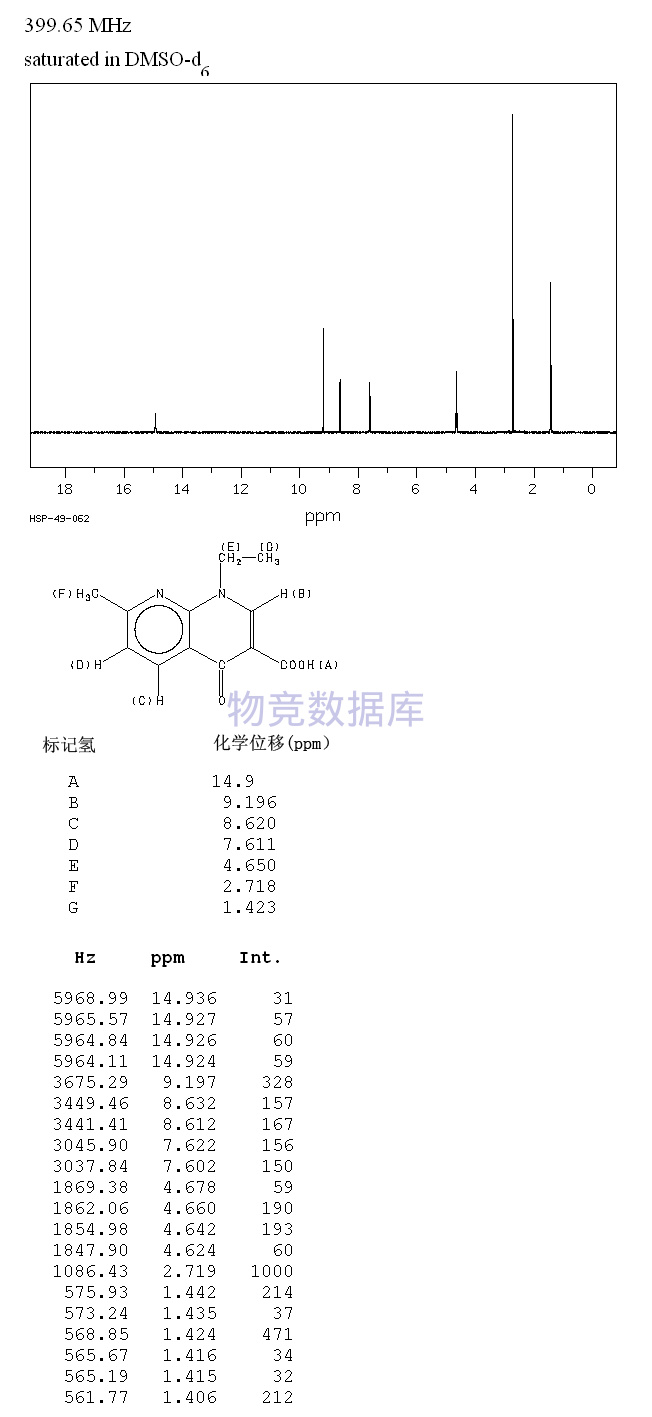
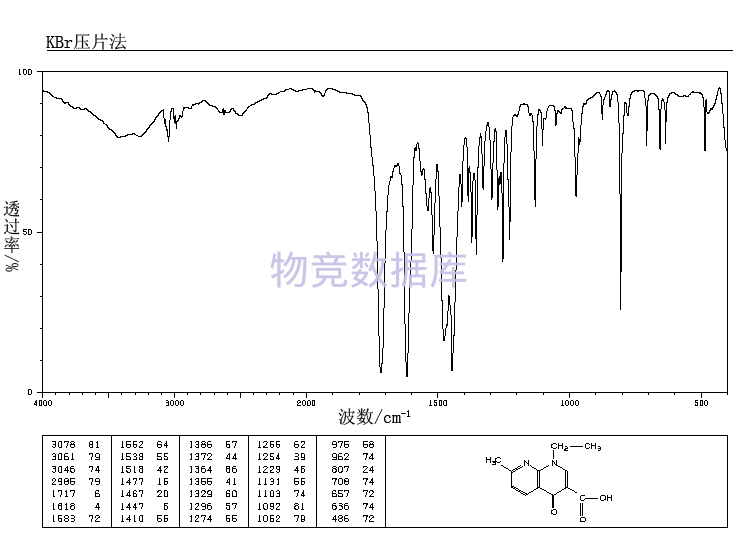
表征图谱


快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号