芳香环状低聚体及其应用前景
- 2012-12-06
- 专题
由于航天﹑航空﹑军事工业及高新技术对高性能树脂及先进热塑性复合材料的依赖和需求,因而对它们的研究历来是国家高技术发展计划中有关新材料领域中的重要组成部分。人们在经历了数十年对高性能树脂的研制、发展和应用评价之后才筛选出为数不多的具有重要商业价值的高聚物,如聚醚醚酮(PEEK)、聚醚砜(PES)、聚酰亚胺(PI)及聚苯硫醚(PPS)等。然而这类高性能树脂在加工成型和复合材料制备工艺上遇到了麻烦。此外,它们对新的高分子加工工艺如增强反应注射成型(RRIM)及树脂传递模型(RTM)亦难以适应,这是由于经逐步聚合反应制备的这类线性高分子量树脂的合成方法及树脂的熔体粘度所造成的。一种由芳香环状预聚体(aromatic cyclic oligomer)的开环聚合来制备高性能树脂的方法为高性能树脂的现代加工工艺及高性能树脂的热塑性复合材料的制备带来了革命性的变革[1,2]
1 芳香环状低聚体
芳香环状低聚体是指在主链上含有很少的脂肪链或不含脂肪链的全芳香的聚碳酸酯、聚酯、聚醚、聚硫醚、聚酰胺及聚酰亚胺等环状同系物(结构式见图1,X和Y代表酯键﹑醚键等链段,n代表重复单元数),它也是继卟啉、杯芳烃、环糊精、冠醚之后的新一类环状结构的化合物。

长期以来,当人们在合成线性高分子量芳香聚合物的过程中就已发现有环状低聚物的存在[3,4],它作为聚合反应的副产物,影响着聚合物产物的性能和应用。有关芳香环状物的报道主要集中在确认和分离存在于线性高聚物中的环化物的工作上。最早有目的地合成芳香环状低聚体始于1962年,Bottenbruch[5,6]、Prochaska[7,8]和Moody[9]等先后报道了在高稀释溶液中七元和八元环状聚碳酸酯以及双酚A 环状聚碳酸酯的合成与开环聚合,然而产率较低,最高化为21% 。此后有关芳香环状低聚体的报道很少。直到美国G. E .公司Brunelle[1,2]领导的研究小组公布了利用“假高稀”(pseudo high dilution)(又称“拟高稀”)技术合成芳香环状聚碳酸酯并成功地进行开环聚合这一具有革命性意义的技术以来,芳香环状低聚体的研究开始成为高分子化学领域的研究热点,进入了高速发展的新阶段。
G. E .公司所进行的开拓性研究为芳香环状低聚体的发展作出的贡献主要体现在两个方面:(1)提出了通过开环聚合来制备热塑性树脂材料的新构思。随着航空、航天事业的发展,对先进复合材料的要求越来越高,开环聚合这一构思恰恰顺应了时代的需求,推动了芳香大环化合物的前进。(2)把“假高稀”技术运用到环状低聚体的制备中来,使得成环反应成为有价值的聚合反应,环状化合物的产率大大提高成为可能。
“假高稀”技术是一种通过控制反应溶液浓度的方法营造出高稀反应条件的一种技术。由于成环反应为不可逆反应,把反应物配成较浓的溶液,然后将反应物溶液缓慢滴加到反应体系中,随着环状低聚物的不断生成,使反应物的未反应端基保持在极低的浓度。通过控制滴加速度可以控制反应物的浓度,避免了大量溶剂的使用。“假高稀”技术最早由Ziegler[10]在1933年提出,但在环状低聚物的合成过程中才得到充分的运用,近几年的研究和探索已经证明“假高稀”技术是由单体一步合成环化物的必要手段,并已成功地合成出多种结构的环状化合物。
继G. E .公司[11,12]合成双酚A 环状聚碳酸酯并研究其成环和开环聚合机理之后,人们运用各种聚合反应,先后成功地合成了大量的芳香环状聚酯和芳香环状聚醚。例如,聚酯方面,Brunelle[13]和Gibson[14]等分别以间苯二甲酰氯和对苯二甲酰氯为单体得到双酚A 环状聚酯;chen等[15]以邻苯二甲酰氯为单体,高产率地合成了双酚A及酚酞等九种环状聚酯;Brittain等[16]使用易于成环的柔性单体与对萘二甲酰氯反应制得含萘基的环状聚酯;Miyano等[17]制备了带有手性联萘基团的芳香环状聚酯。聚醚方面,Colquhoun等[18]率先报道了单分散全芳香环状聚醚的合成;为降低所得环状低聚体的熔点,Gibson等[19]合成了一系列含不对称结构的芳香环状聚醚;Hay等[20,21]接连报道了一系列含1,2-二苯甲酰基苯基团的环状聚醚的合成、聚合、对其流变学的研究以及一系列可交联的含炔基的环状聚醚[22]和具有阻燃性的含膦酰基团的环状聚醚[23]的合成;Gibson等[24]通过多步合成法合成了性能优异、用途广泛的聚醚醚酮(PEEK)和聚醚酮(PEK) 。
芳香环状聚硫醚的一个重要特点是可以进行自由基开环聚合,能够避免聚合产物中含有阴离子引发剂的端基,故比阴离子开环聚合更具有应用价值。Hay等深入研究了含吸电子基团1,2-二苯甲酰基苯的环状聚硫醚酮低聚物[25],合成了一系列新型的芳香环状聚硫醚醚低聚物和含过硫键的芳香环状低聚物[26,27],另外还以4-溴代苯硫酚铜为单体高选择性地合成了聚苯硫醚(PPS)环状低聚物[28]。环状聚酰亚胺的合成一般采用长链的柔性二酐或二胺单体[29,30];相对于其它芳香环状低聚体,全芳香的环状聚酰亚胺由于分子结构刚性较强更加难以合成,但由于它具有一些特殊性能,最近也开始引起人们的关注[31]。
芳香环状低聚体的合成主要受单体浓度、溶剂和催化剂、化学计量平衡、单体的分子结构等因素的影响。需要指出的是单体的分子结构对环化反应起着重要的作用:如果单体的构型不适合成环,那么无论如何优化其它反应条件都难以得到高产率的环化产物。分子结构不仅影响成环率的高低,也影响环状低聚体的尺寸和分布。对于芳香体系,单体的中心键角在 90~180°范围内,其键角小于109°时,分子易于成环;大于120°时,生成线性聚合物的倾向增加[32]。为了精确地表征芳香环状低聚体的结构,经常使用多种测试手段并用的综合表征法,包括NMR、GPC、HPLC、MALDI-TOF-MS等;有时辅助的分析手段也是不可缺少的,如DSC、X 射线衍射等方法。
与线性高聚物及其它小分子环状化合物相比,芳香环状低聚体在分子结构和性能上的特点为:(1)由一系列聚合度不同的环状同系物组成,具有一定的组分分布;熔融粘度低;通常具有较大的分子尺寸,缺乏环张力;因此,在引发剂引发下,能够进行进一步的开环聚合,并且在聚合过程中没有小分子量挥发性副产物生成。(2)具有刚性和较为敞开的分子结构,且具有一定的空穴,这是其在分子识别及其他一系列应用方面的结构基础。(3)具有很高的热稳定性,在有机溶剂中溶解性好,利于进行加工和改性。
2 芳香环状低聚体的应用
以开环聚合为应用目的的芳香环状低聚体自兴起至今的十多年的研究发展表明,芳香环状低聚体不仅仅适用于开环聚合,结构和性能上的优点决定了其更为广泛的应用领域。芳香环状化合物在许多前沿领域均有诱人的潜在的应用前景。
2.1用于熵驱动的开环聚合(ED-ROP)
由芳香环状中间体的开环聚合(图2)制备高性能树脂在商业上极具吸引力:(1)开环聚合反应把低分子量、低粘度的环状预聚体转化成高分子量聚合物而不形成任何副产物;(2)操作这些低粘度预聚体的加工过程比高粘度的高分子量聚合物要容易得多;(3)低粘度加上不形成小分子副产物,对碳纤维或玻璃纤维增强的复合材料的制备具有重要的价值;(4)避免在模压或注射成型中遇到的部件的应力集中等问题。由于具有较大的环尺寸和较小(或不存在)的环张力,芳香环状齐聚物的开环聚合属于由熵驱动的聚合反应,反应过程中无副产物和反应热生成,在引发剂引发下,低粘度的环状预聚体直接转化成高分子量聚合物。这些特点使其在聚合加工如增强反应注射成型(RRIM)和树脂转移模塑(RTM) 方面具有重要价值。迄今为止,已有百余种芳香环状低聚体经开环聚合得到了相应的高分子量的线性聚合物。ED-ROP 的典型实例如G. E .公司Brunelle等对环状双酚A 碳酸酯的开环聚合,以有机钛酸酯为交换引发剂,在300℃下加热30min,环状碳酸酯发生开环聚合,得到已商品化的分子量高达2.5×105的线性聚碳酸酯[33]。

开环聚合产物的分子量普遍偏低,这是一个应用上的瓶颈问题,亟需开发新的聚合方法和新型引发剂以提高聚合产物的分子量。
2.2用于分子识别
很多识别系统包含环状化合物。分子识别即指主体(受体)对客体(底物)选择性结合并产生某种特定功能的过程,它们主要依靠称为非共价键力的分子间的作用力(包括范德华力,疏水相互作用,氢键等)。而芳香环状低聚体的主链中多带有酯基﹑醚基﹑酰胺基等基团,这些基团不仅能够形成分子内的氢键作用,也能与其它分子形成分子间的氢键作用,这种较强的氢键作用与芳环之间的π-π作用等分子间力是其用于识别系统的基础[34]。例如,图3(Ⅰ)所示结构的“分子盒”能够识别乙炔基或氰基[35];Inouye等设计并合成了一种特殊结构的芳香环状酰亚胺,该环状低聚物可识别呋喃核糖分子[36](图3,Ⅱ);Hunter等合成的一种新型索烃(图3,Ⅲ)可识别苯醌[37]。

很多芳香环化物由于结构上的特点也具有识别能力。随着大量制备芳香环状低聚体手段的出现,这一领域的研究有望得到快速发展。
2.3用于高分子纳米复合材料的制备
纳米技术是21 世纪最受瞩目的三大高新技术之一。将高分子与层状粘土﹑石墨等无机填料相结合制备成高聚物/无机纳米复合材料,由于在纳米相与基体间形成特殊的结构,能够大大提高聚合物性能并拓展其应用,因而成为科学研究的热点。制备高聚物/ 无机纳米复合材料通常采用聚合物熔体插层法。但对于由芳香族聚合物制备纳米复合材料,由于芳香高聚物熔点较高,而长时间在熔点状态下又会引起高聚物的热降解,故此法不可行。而由芳香环状预聚体的原位开环聚合则可有效地解决这一难题。例如,Hay等[38]将环状预聚体与纳米级层状粘土充分混合,通过扩散﹑极性基团作用﹑微孔吸附等使预聚体充分进入粘土片层间,不需引发剂,在 150℃下进行开环聚合,不产生任何副产物或挥发性气体,片状粘土便以纳米尺寸均匀分散在高聚物基体中,得到芳香聚硫醚/ 粘土纳米复合材料。Hay[39]还制备了芳香聚硫醚/ 石墨纳米复合材料,将环状聚芳硫醚与具有优良导电性的片层石墨(重量比15%)充分混合,开环聚合后即得到导电高分子纳米复合材料。
高聚物/无机纳米复合材料兼具组分中无机纳米粒子和高聚物的特点,并将无机物的刚性﹑尺寸稳定性和热稳定性与聚合物的韧性﹑易加工性等完美地结合起来,性能优异,可广泛用于各个领域,在交通工具﹑飞机部件﹑耐油容器﹑油箱及电气﹑电子﹑光电产品等方面都有较为广阔的应用前景。
2.4用于其它功能材料的研究
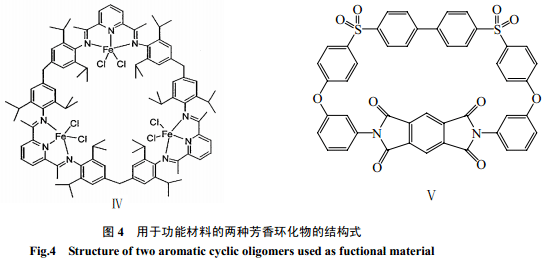
由于芳香环状化合物特有的结构及高热稳定性,它在多个研究方向上均引起人们的高度关注。最近李悦生等将铁乙烯聚合催化剂负载在大环化合物上,首次制备出多核环状后过渡金属烯烃聚合催化剂[40],与单核吡啶双亚胺铁催化剂相比,环化物负载催化剂展示出更高的活性和更长的催化剂寿命(图4,Ⅳ)。Colquhoun等[31]以大约20%的收率合成了大环芳香聚醚-酰亚胺-砜聚体(图4,Ⅴ),研究了它们的配位性能,发现这类大环的络合能力很强,并且得到的配合物具有非常高的热化学稳定性,并预言这类大环分子在未来超分子化学的发展中占据重要地位。Colquhoun等[41]还通过芳香大环预聚体的熔融开环聚合得到了尺寸在200~400nm 的高性能芳香聚合物的纳米管和纳米纤维。Miyashita等[42]通过含有螺旋状手性基团的单体合成了一系列具有旋光性的芳香大环聚酰胺,制备出一种新型LB膜,即手性LB膜,在手性鉴别和手性传感设备上具有光明的前景。张万金等[43]利用纳米尺寸的刚性环状聚醚醚酮酮(PEEKK)齐聚物,制备了稳定的二维有序LB膜,在药物控制和光化学太阳能转化方面有望获得应用。
此外,芳香环状化合物可在分子中引入各种官能团(如氨基、羧基、酸酐等),通过这些官能团的反应,将对开发适用于可控交联、分子海绵结构和精密电子器件等领域的新型材料产生深远影响;利用芳香环状化合物良好的热稳定性和敞开式的分子结构的特点,它在环线互穿网络(polyrotaxanes or catenanes) 方面亦有潜在的应用[44]。大量研究结果说明,芳香环状低聚体在功能材料方面具有多种用途,因此继续这方面的研究具有十分重要的意义。
21 世纪是空间技术飞速发展的时代,这对高强度、耐高温、耐冲击的高分子及其复合材料提出了更高的要求。突破传统观念,利用高性能的芳香大环化合物的开环聚合技术为先进树脂基复合材料带来了革命性的变革。今后,芳香环状低聚体的开环聚合将在聚合技术中占据越来越重要的位置。芳香环状低聚体还将广泛应用于超分子化学(包括分子识别)、高聚物纳米复合材料及其它功能材料等领域。随着人们认识的更加深入,芳香环状低聚体将应用于各个方面,开发其潜在的应用价值将成为未来发展的主流,吸引越来越多的科研工作者的视线。
参考文献
[1] D J Brunelle. Ring-Opening Polymerization. New York: Hanser, 1993: 309~361.
[2] D J Brunelle, J E Bradt. USP: 5039783,1991.
[3] C E Beer. J. Polym. Sci., 1955, 13:591~594.
[4] K Tyuzyo, Y Harada, J Suzuki. Polym. Lett., 1964, 2:43~45.
[5] H Schnell, L Bottenbruch. Makromol. Chem., 1962, 57: 1~4.
[6] H Schnell, L Bottenbruch. USP: 3386954, 1968.
[7] R J Prochaska. USP: 3274214, 1966.
[8] R J Prochaska. USP: 3422119, 1969.
[9] L S Moody. USP: 3155683, 1964.
[10] K Ziegler, H Eberle. Chem., 1933, 504: 94~99.
[11] D J Brunelle, E P Boden, T G Shannon. J. Am. Chem. Soc., 1990, 112: 2399~2402.
[12] T L Evans, C B Berman, T C Carpenter et al. Polym. Prepr., 1989, 30(2): 573~574.
[13] T L Guggenheim, S J McCormick, J J Kelly et al. Polym. Prepr., 1989, 30(2): 579~580.
[14] H W Gibson, S Ganguly, N Yamaguchi et al. Polym. Prepr., 1993, 34(1): 576~577.
[15] H Y Jiang, T L Chen, T X Liu et al. Polymer, 1996, 37(15): 3427~3429.
[16] P Hubbard, W J Brittain. Macromolecules, 1996, 29: 8304~8307.
[17] S Miyano, S Handa, M Tobita et al. Bull. Chem. Soc. Jpn., 1986, 59: 235~238.
[18] H M Colquhoun, C C Dudman, M Thomas et al. J. Chem. Soc., Chem. Commun., 1990: 336~338.
[19] D H Xie, Q Ji, H W Gibson. Macromolecules, 1997, 30: 4814~4827.
[20] Y Ding, A S Hay. Macromolecules, 1996, 29: 3090~3095.
[21] Y F Wang, M Paventi, K P Chan et al. J. Polym. Sci., Part A: Polym. Chem., 1996, 34: 2135~2148.
[22] Y F Wang, M Paventi, A S Hay. Macromolecules, 1997, 30: 469~478.
[23] Y Ding, A S Hay. J. Polym. Sci., Part A: Polym. Chem., 1998, 36: 519~526.
[24] [M F Chen, H W Gibson. Macromolecules, 1996, 29: 5502~5504.
[25] K P Chan, Y F Wang, A S Hay. Macromolecules, 1995, 28: 653~655.
[26] Y F Wang, A S Hay. Macromolecules, 1997, 30: 182~193.
[27] Y Ding, A S Hay. Macromolecules, 1996, 29: 6386~6392.
[28] Y F Wang, A S Hay. Macromolecules, 1996, 29, 5050~5053.
[29] J Jazwinski, A J Blacker, J M Lehn et al. Tetrahed. Lett., 1987, 28(48): 6057~6060.
[30] T L Guggenheim, S J McCormick, J W Guiles et al. Polym. Prepr., 1983, 24(2): 138~139.
[31] H M Colquhoun, D J Williams, Z Zhu. J. Am. Chem. Soc., 2002, 124(45): 13346~13347.
[32] M F Teaslay, D Q Wu, R L Harlow. Macromolecules, 1998, 31(7): 2064~2074.
[33] D J Brunelle, T G Shannon. Macromolecules, 1991, 24: 3035~3044.
[34] A J Hall, P Hodge. Reactive & Fuctional Polymers, 1999, 41: 133~139.
[35] I Baxter, H M Colquhoun, P Hodge et al. Chem. Commun., 1998: 283~284.
[36] M Inouye, T Miyake, M Furusyo et al. J. Am. Chem. Soc., 1995, 117: 12416~12425.
[37] C A Hunter. J. Chem. Soc., Chem. Commun., 1991: 749~750.
[38] X S Du, M Xiao, A S Hay et al. Polym. Int., 2004, 53: 789~793 .
[39] X S Du, M Xiao, A S Hay et al. Polymer, 2004, 45(19): 6713~6718.
[40] J Y Liu, Y S Li, Z S Li. Macromolecules, 2005, 38(7): 2559~2563.
[41] H M Colquhoun, M G Zolotukhin, L G Sestiaa et al. J. Mater. Chem., 2003, 13: 1504~1506.
[42] F Feng, T Miyashita, H Okubo et al. J. Am. Chem. Soc., 1998, 120: 10166~10170.
[43] T Ben, C H Chen, W J Zhang et al. Macromol. Rapid Commun., 2002, 23: 196~199.
[44] S H Lee, H W Gibson. Macromolecules, 1997, 30: 5557~5559.
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号