烯类单体在室温离子液体中的自由基聚合反应研究进展
- 2012-12-07
- 专题
室温离子液体是一类在室温(或低于100℃)呈熔融状态的盐, 通常由体积较大的有机阳离子和无机酸根离子组成[1,2]。与常见的有机溶剂相比, 有如下一些特点:(1)蒸汽压低、挥发性弱, 除了能满足一般化学反应和分离提纯操作的需要外, 还适用于要求高真空或较高温度的反应体系;(2)稳定性好, 不易燃、易爆;(3)溶解能力易调, 容易回收利用;(4)极性较高、络合能力较弱, 是使用过渡金属催化剂反应的理想介质;(5)对某些反应有一定的催化和选择作用。基于这些独特的性质, 室温离子液体被认为是继超临界CO2之后的新一代“绿色”溶剂, 相关研究十分活跃。
关于离子液体的合成、性质和应用, Seddon[1]、Welton[2]等已有详细的评介。自由基聚合反应涉及的聚合物量大、面广, 本文将主要介绍烯类单体在室温离子液体中自由基聚合反应的研究情况, 并结合笔者正在进行研究进展, 提出对今后工作的粗浅看法。
1室温离子液体中的自由基聚合反应
1. 1普通自由基聚合反应
Hong等[3,4]以偶氮二异丁腈(AIBN)和过氧化苯甲酰(BPO)为引发剂, 分别研究了甲基丙烯酸甲酯(MMA)和苯乙烯(St)在1-丁基-3-甲基咪唑六氟磷酸盐([bmim][PF6])和苯中的自由基聚合反应。发现在离子液体中聚合得到的聚甲基丙烯酸甲酯(PMMA)和聚苯乙烯(PSt)与在苯中得到的相应聚合物具有相似的分子量分布, 但在离子液体中的聚合速度快, 聚合物分子量比在苯中聚合得到的大约10倍。他们认为主要原因可能有二:一是聚合过程中聚合物析出, 增长链末端的自由基由于被高分子链包埋而减少了链转移和终止的机会;另一是离子液体的粘度比苯高, 增长链自由基通过扩散会聚而发生碰撞的几率比在苯中的小。这两个原因都可使自由基的寿命延长, 聚合速度增加。但后来的研究表明, 虽然PSt在[bmim][PF6]中不溶, 但PMMA 在[bmim][PF6]中是可溶的[5~7],MMA在[bmim][PF6]中聚合时反应体系一直保持均相。因此, 前述的第一个原因不能解释MMA在[bmim][PF6]中聚合速度快、聚合物分子量大的事实。
Harrisson等[5~6]采用脉冲激光聚合(pulsed laser polymeriztion)技术深入地研究了MMA在离子液体中链增长速率常数(kp)与离子液体浓度的关系。结果表明, kp随离子液体浓度的提高而增加, 最高可超过本体中的2倍, 远远高于一般有机溶剂对kp的影响。虽然尚不完全清楚为什么离子液体具有这么明显的溶剂诱导加速聚合作用, 但可部分解释为什么MMA在[bmim][PF6 ]中聚合速度快、聚合物分子量大。
普通自由基聚合反应一般不能用来合成嵌段共聚物。利用离子液体中增长链自由基寿命较长的特点, Zhang等[8]通过顺序加料的方法, 用自由基聚合制备了聚苯乙烯和聚甲基丙烯酸甲酯嵌段聚合物。使用的离子液体是[bmim] [PF6],引发剂是BPO, 反应温度为70℃。先合成PSt链段, 聚合到一定程度后减压将未反应的St抽出, 再加入第二单体MMA于室温聚合, 即可得到PSt-b-PMMA 嵌段共聚物, 并经1HNMR、DSC和分级沉淀等方法证实。
溶剂的回收利用是所有溶液聚合都必须面对的问题, 因为残存的溶剂不仅会造成环境污染, 而且会严重影响高分子制品的最终性能。离子液体蒸气压低、不易挥发, 不能用常见的加热或抽真空的方法除去, 必须采用其它的方法。Benton等[9]研究了离子液体的回收利用问题。聚合完成后, 用V(乙醇)/V(水)= 80/20的混合溶剂萃取含有[bmim][PF6]的PMMA 薄膜。在23℃时, 如果时间足够长, 最终有97%的离子液体被萃取出来; 而在55℃时, 则萃取速度要快得多, 2h内可萃取出87%的离子液体。旋干萃取液, 即可回收离子液体。
残留的少量离子液体可让其保留在聚合物中, 起增塑剂的作用。Scott等[10]发现[bmim] [PF6]是PMMA 的有效增塑剂。当聚合物中离子液体的重量浓度达到50%时仍然是均相体系, 而当常用的增塑剂邻苯二甲酸二辛酯的浓度达到40%~50%时已有明显的相分离发生。此外, 以该离子液体为增塑剂的聚合物力学性能与用邻苯二甲酸二辛酯为增塑剂的相当, 但热稳定性更好。
1. 2活性自由基聚合反应
1. 2. 1ATRP聚合反应 原子转移自由基聚合反应(ATRP)是迅速发展的一种活性自由基聚合技术[11], 由有机卤化物(引发剂)、过渡金属低价态盐(催化剂)及含氮多齿配体构成引发体系, 反应机理如图1所示。聚合过程中, 低价金属离子从引发剂分子夺取一个卤原子后变成高价金属离子,而引发剂分子失去卤原子后生成自由基, 该自由基向单体加成诱导聚合反应。增长链自由基也可以从高价态过渡金属盐夺取一个卤原子后失去活性, 生成休眠种; 同时, 将金属盐从高价态还原为低价态。引发剂的“活化”与“失活”是可逆反应, 速度很快, 反应体系中活性中心的浓度远较普通自由基聚合体系中的低, 并且保持恒定, 从而有效调控聚合反应的进行。
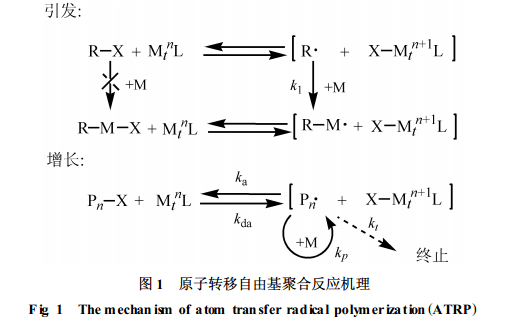
从上述的反应机理可以看出, 反应体系中过渡金属化合物的含量对于聚合反应的控制起着决定性的作用。普通有机溶剂对过渡金属化合物的溶解性差, 在其中进行的ATRP一般都是非均相反应。为了达到对聚合反应有效控制的目的, 往往需要加入较大量的金属催化剂, 这一方面增加了反应成本, 另一方面又不可避免地造成残留金属离子对聚合物的污染。为了解决这—问题,Carmichael等[7]开展了MMA在[bmim][PF6]中进行ATRP反应的尝试, 如图2所示。所用的引发剂是2-溴异丁酸乙酯, 催化剂是CuBr, 有机配体是N-丙基-2-吡啶基甲亚胺(N-proyl-2-pyridylmethanimine)。该聚合反应具有反应速度快、反应温度低(低至30℃)、聚合物分子量分布窄的特点。由于[bmim][PF6 ]与甲苯不相溶, 反应完成后可用甲苯从反应混合物中将聚合物萃取出来, 而绝大多数金属离子仍留在离子液体中, 不会污染聚合物。

Sarbu等[12,13]合成了一系列阳离子为1-丁基-3-甲基咪唑鎓、阴离子分别为Cl-、Br-、A lCl4-、CO32-、(C4H9)2PO4-的离子液体, 系统地研究了阴离子对MMA在其中的ATRP反应的影响。当以2-溴异丁酸乙酯为引发剂, 在催化剂量的离子液体存在下,MMA在本体、甲苯或苯甲醚等溶剂中可以进行ATRP反应。如果以Fe(Ⅱ)为催化剂, 不使用配体也可调控MMA的活性聚合反应;而如果以Cu(Ⅰ)为催化剂, 则只有当[bmim][(C4H9)2PO4]存在时才可不使用配体。含有过渡金属盐的离子液体很容易与非极性的聚合物溶液分离, 催化剂的回收和循环使用也可以方便地解决。
Biedron等[14]报道了一系列丙烯酸酯单体在[bmim][PF6]中的ATRP聚合反应。单体在[bmim][PF6]中的溶解性取决于单体分子上烷基的长度。丙烯酸甲酯(MA)在[bmim] [PF6]中的反应为均相反应, 所得聚合物分子量的实测值与计算值接近, 且分子量分布较窄。丙烯酸丁酯(BA)虽然不溶于[bmim][PF6],为非均相聚合反应, 但当搅拌速度足够快能够保证引发剂、单体和增长链自由基在两相之间的转移时, 聚合反应仍以可控的方式进行。而对丙烯酸己酯和丙烯酸十二烷基酯, 即使施以充分搅拌, 反应仍然不可控。
基于前面的工作,Biedron等[15]又研究了丙烯酸酯类单体在离子液体中的嵌段共聚合反应。在[bmim][PF6]中, 先用ATRP方法合成聚丙烯酸丁酯大分子引发剂, 在BA几乎完全聚合后, 直接加入第二种单体MA, 所得嵌段聚合物分子量分布很窄, 均聚物含量很少, 数均分子量比理论值稍高。反之, 如果先在离子液体中合成PMA 作为大分子引发剂, 在MA转化率低于70%时加入第二单体BA, 仍可得到分布较窄的嵌段聚合物。但是, 当转化率高于70%时再加BA, 聚合产物中除了嵌段共聚物外还有均聚物。而当转化率大于90%时再加BA, 则根本得不到嵌段共聚物。端基分析表明, 在均相体系中, 当MA的转化率高于70%时, 增长链自由基会发生不可逆终止; 而在非均相体系中, 增长链自由基不可逆终止的速率要小得多。
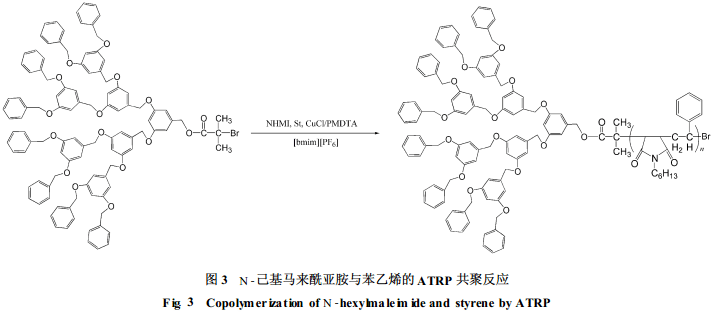
习复等[16]以含溴端基、不同代数的树枝状分子Dendriticpolyacrylether 2-bromoisobutyra te为引发剂, 用ATRP方法合成了N-己基马来酰亚胺与苯乙烯共聚物, 如图3所示。该反应的引发效率很高(99%), 反应温度低(30℃), 所得聚合物的分子量分布较窄(1.18<Mw/Mn<1.36), 受催化剂的污染较少。与在普通有机溶剂如苯甲醚中进行的反应相比, 在较大的投料比范围内, 在离子液体中更易形成N-己基马来酰亚胺和苯乙烯的交替共聚物。
1.2.2 反向ATRP聚合反应
反向ATRP与ATRP一样, 也是借助过渡金属催化剂调控的氧化还原反应降低反应体系中增长链自由基的浓度, 实现对聚合反应的控制。在ATRP中, 引发剂是烷基卤化物, 催化剂是过渡金属的低价盐。前者对人体和环境的危害较大, 后者使用时易被氧化。为了克服ATRP的这两个缺点,Matyiaszewski等[17]提出了反向ATRP的概念。其主要特点是用AIBN、BPO等普通自由基聚合引发剂代替烷基卤化物, 用过渡金属高价盐代替低价盐, 从而有效解决了普通ATRP中引发剂的毒性和催化剂的易氧化性问题。
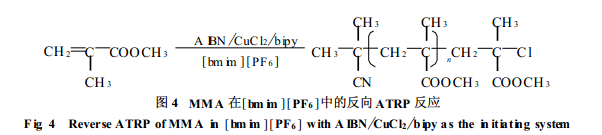
笔者研究了MMA在[bmim][PF6]中的反向ATRP反应, 采用的引发体系是AIBN/CuC l2/bipy(联吡啶), 如图4所示。Matyjaszewski等曾报道AIBN/CuCl2/bipy引发体系不能有效调控MMA的自由基聚合反应, 主要原因是催化剂在本体中或其它有机溶剂中溶解性较差, 为非均相聚合体系。同时, 单体MMA的反应活活较高, 使体系中自由基浓度过高而无法控制。而当以4, 4’-二(5-壬基)-2, 2’-联吡啶(dNbipy)为配体、苯甲醚为溶剂形成均相体系时, 反应则可控[18]。笔者利用离子液体对于金属化合物良好的溶解性, 选用[bmim][PF6]作为聚合反应的溶剂, 可形成均相反向ATRP聚合体系[19]。通过对不同温度下聚合反应动力学的研究以及对聚合物端基的分析, 确定该聚合反应为活性聚合, 所得聚合物的分子量分布很窄(Mw/Mn<1.2)。另外, 由于催化剂在[bmim][PF6]中的良好溶解性, 只需加入少量催化剂([AIBN][CuCl2]= 8/1)即可有效调控聚合反应, 从而减少了金属离子对聚合物的污染。从反应混合物中分离聚合物后, 仅需经过简单处理, 离子液体即可回收并重复使用。再向回收的离子液体中加入MMA和AIBN, 反应又可以同样的方式进行。
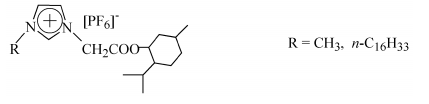
以上工作是在疏水性离子液体中完成的, 笔者还进行了MMA在亲水性离子液体[C4mim][BF4]和[C12mim][BF4]中的反向ATRP反应[20]。文献报道含BF4-的离子液体对过渡金属化合物有更好的溶解性[21], 因而希望在其中的自由基聚合反应可以得到更好的控制。为了进一步研究聚合时手性环境对聚合物立构规整性的影响, 笔者设计合成了一类含有不对称中心的离子液体[22], 化学结构如下:
由于离子液体对催化体系具有很好的溶解性, 仅向反应体系中加入少量的手性离子液体m(MMA)/m(ILs)=3/1即可调控MMA的反向ATRP聚合反应, 获得分子量分布很窄的聚合物(Mw/Mn<1.2)。以所得的聚合物作为大分子引发剂, 可引发甲基丙烯酸薄荷醇酯(MnMA)的ATRP反应, 获得具有光学活性的嵌段聚合物。
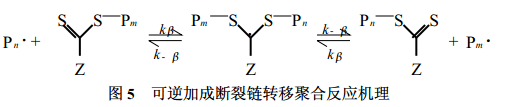
1. 2. 3 RA F T聚合反应 可逆加成断裂链转移聚合(RAFT)是新近发展起来、适用单体范围更广的一种活性聚合技术, 其反应机理如图5所示[23]。与普通自由基聚合反应相比, 体系中除了单体、引发剂外, 还有调控增长链自由基浓度的二硫代羰基化合物。聚合时, 引发剂引发单体聚合形成增长链自由基, 增长链自由基向二硫代羰基化合物的CS 键可逆加成形成自由基中间体, 该中间体可逆断裂又形成新的增长链自由基。由于在反应过程中, 自由基的浓度远比二硫代羰基化合物的浓度低, 链终止和链转移的程度降低, 从而可达到控制聚合反应的目的。
Perrier等研究了AIBN引发的MMA、MA和St在离子液体中的RAFT聚合反应[24]。他们选用的二硫代羰基化合物和离子液体的化学结构为:
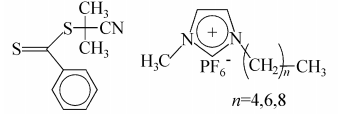
聚苯乙烯不溶于所用的离子液体, 在反应的初期就沉淀出来, 基本得不到聚合物。MMA、MA及其聚合物在三种液体中都有很好的溶解性, 聚合以活性的方式进行, 所得聚合物的分子量分布(PDI)较窄。对PMMA, PD I值小于1. 1; 而对PMA, PDI值虽然比PMMA 的大, 但仍小于1.3。在离子液体中的聚合速度比在本体和其它溶剂中的大, 并且和离子液体的结构有关。在其它条件完全相同的情况下,M M A在[C4mim] [PF6]中的转化率是84. 3% ,而在[C6mim] [PF6]和[C8mim] [PF6]中的转化率分别为91.3%和90.1%。所得聚合物的末端基确定, 可与新加入的单体进行扩链反应。
2 展望
Seddon[25]曾指出, 几乎所有类型的有机反应都可以在离子液体中进行。现在, 大量的实验事实已经证明了他的预言。离子液体所提供的反应环境与传统的溶剂有很大差别, 从而产生了许多新反应、新现象、新结构和新功能, 并且有可能减少污染, 保护环境。可以说, 离子液体为化学科学和化学工业的发展提供了新的机遇。相比之下, 离子液体中烯类单体的自由基聚合反应研究开展得还很少, 有许多尚待探索的领域, 今后的工作可能包括新型离子液体的设计、合成、表征以及离子液体中的自由基聚合反应规律、活性自由基聚合反应、立构规整自由基聚合反应等方面。其中, 功能性离子液体的设计、合成及在烯类单体自由基聚合反应中的应用将受到进一步的关注。功能性离子液体是指既可作为反应介质, 又具有催化、分子识别、络合等功能的离子液体, 如离子液晶、手性离子液体、带有配位基团的离子液体等。它们可以提供不同于一般有机溶剂的反应环境, 从而有助于在更高水平上实现对聚合反应和高分子结构的控制。此外, 利用离子液体合成其它类型的高分子已有一些报道, 毫无疑问这也将是今后发展的重要方向。
参 考 文 献
[1] KRSeddon. J. Chem. , T ech. B io techno l. , 1997, 68: 351~356.
[2] TW elton. Chem. R ev. , 1999, 99: 2071~2083.
[3] HWZhang, KLHong, J MM ay s. Po lym. P rep. , 2001, 42: 583~584.
[4] KLHong, HWZhang, J MM ay seta l. Chem. Comm un. , 2002, 13: 1368~1369.
[5] SH a rrisson, SRM ackenze, DMH add leton. Po lym. P rep. , 2002, 43: 883~884.
[6] SH a rrisson, SRM ackenzie, DMH add leton. Chem. Comm un. , 2002, 23: 2850~2851.
[7] AJ Ca rm ichael, DMH add leton, SAFBoneta l. Chem. Comm un. , 2000, 14: 1237~1238.
[8] HWZhang, KLHong, J WM ay s. M acrom o lecu les, 2002, 35: 5738~5741.
[9] MGB en ton, CSB razel. Po lym. P rep. , 2002, 43: 881~882.
[10] MPSco t t, CSB razel, MGB en toneta l. Chem. Comm un. , 2002, 13: 1370~1371.
[11] J SW ang, KM a ty ja szew sk i. J. Am. Chem. Soc. , 1995, 117: 5614~5615.
[12] TSa rbu, KM a ty ja szew sk i. Po lym. P rep. , 2002, 43: 211~212.
[13] TSa rbu, KM a ty ja szew sk i. M acrom o l. Chem. Phy s. , 2001, 202: 3379~3391.
[14] TB ied ro, PKub isa. M acrom o l. R ap idComm un. , 2001, 22: 1237~1242.
[15] TB ied ro, PKub isa. J. Po lym. Sci. Po lym. Chem. , 2002, 40: 2799~2809.
[16] YL Zhao, JMZhang, J J iang eta l. J. Po lym. Sci. Po lym. Chem. , 2002, 40: 3360~3366.
[17] J SW ang, KM a ty ja szew sk i. M acrom o lecu les, 1995, 28: 7572~7573.
[18] J HX ia, KM a ty ja szew sk i. M acrom o lecu les, 1997, 30: 7692~7696.
[19] HYM a, XHW an, XFCheneta l. J. Po lym. Sci. Po lym. Chem. , 2003, 40: 143~151.
[20] HYM a, XHW an, XFCheneta l. Po lym er, 2003, 44: 5311~5316.
[21] LCB ranco, J NRo sa, J JMR am o seta l. Chem2Eu r. J. , 2002, 8: 3671~3677.
[22] HYM a, XHW an, XFCheneta l. Ch ineseJ. Po lym. Sci. , 2003, 21: 265~270.
[23] J Ch iefa ri, YKChong, FE rco leeta l. M acrom o lecu les, 1998, 31: 5559~5562.
[24] SPerrier, TPD avis, AJ Ca rm ichaeleta l. Chem. Comm un. , 2002, 19: 2226~2227.
[25] MF reem an t le. C&EN. , 1999, 4: 23~24.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号