从维他命B12依赖性酶中得到的环境友好型催化剂
- 2012-12-10
- 专题
1.介绍
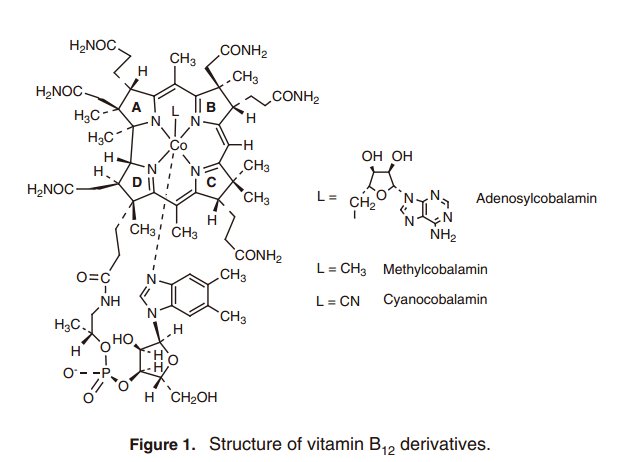
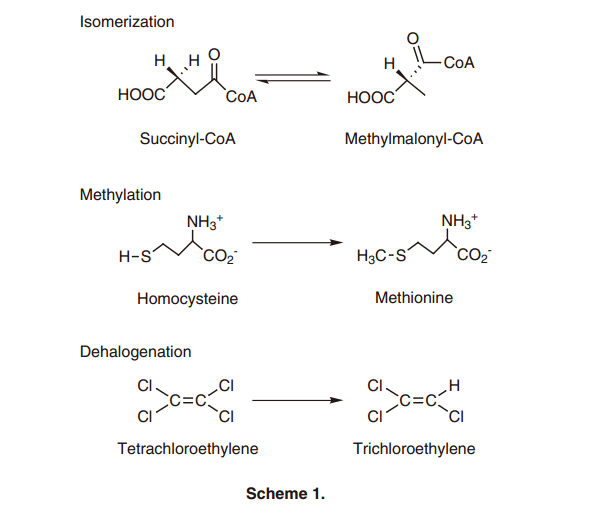
众所周知维他命B12衍生物是一种独特的辅酶,它内部具有钴-碳(Co–C)键,与多种脱辅基蛋白组合参与各种催化功能。1) 维他命B12是一个金属配合物,在咕啉环中钴离子与四个氮原子环相互配合(图1)。它最初用作治疗恶性贫血的神奇武器,它独特的结构是由Hodgkin等人通过X-ray crystallography所描述。2)中央钴离子的氧化价态由+1到+3; Co(I)呈灰绿色, Co(II)呈黄色-橙色,Co(III),是最常见的形式,在B12衍生物中显红色,也被称为“红色维生素。”甲钴胺和腺苷钴胺作为体内酶辅酶形式在生物合成蛋白酸酶和碳骨架异构化重排中分别具有催化功能。3) 最近的研究表明,例如咕啉环复合物结构在厌氧细菌的脱氯反应中起到活性中心的作用;这表明B12依赖酶的一个新的功能(方案1)。4)
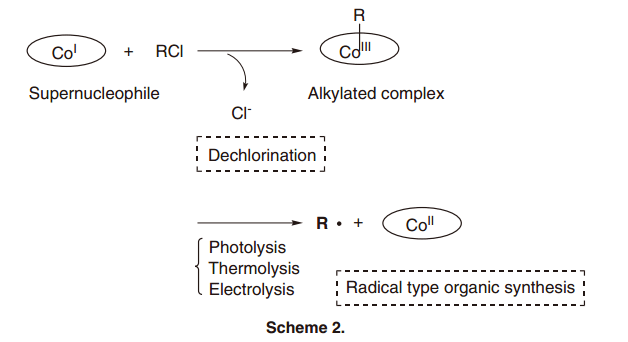
B12依赖酶反应的显著特点是与金属碳键形成了一个独特的有机金属结构。这种结构主要是由Co(I)与有机卤化物,Co(III)与Grignard试剂反应所得, 它很容易在温和的外源刺激,比如说可见光、热、氧化还原条件下发生均裂。特别是, 当前反应发生时,Co-C键的形成包含了有机卤化物的脱卤,并产生有机自由基种在的Co-C键的断裂(方案2)。通过专注于上述Co-C键的特点, 我们研发了一个分子转换,利用Co-C键与B12模型复合物的形成于分裂来激化B12依赖酶的活性中心。5)在这篇文章中, 我们提出了我们的结果:维他命B12衍生物的合成及其催化过程。
2. 维生素B12建模
Vitamin B12从氰钴胺生产生物体重大量获得,用于膳食补充或者是牲畜饲养。目前为止,没有任何关于使用天然维生素B12作为催化剂原料的安全性或者是经济型方面的问题产生。然而, 从细菌中获得的天然维生素B12可能回含有维生素B12相关物质,这表明它有可能会具有较差的化学稳定性。这不是因为咕啉环的结构的不稳定性,而是侧链取代基的退化。正如后面锁描述的, 咕啉环具有很高的稳定性。要使用vitamin B12衍生物作为催化剂, 应满足下列条件:(1)应保留咕啉环结构,(2)中央钴原子应该是完好的, (3)修改的侧链应该维持不变。在这三个条件中, 我们暂时从氰钴胺中移除生理必须的侧链结构,通过所有的外周侧链的化学修饰作用生成酯用于合成疏水性维生素B12(heptametyl cobyrinate) (图2, A).6) 酸催化剂参与下,通过加热醇溶液中的氰钴胺的简单合成,可以获得80%的疏水性vitamin B12,即使氰钴胺被用作原料,它也是通过以上提到的大量的微生物所得到的。使用疏水性维生素B12作为起始原料,它可以合成各种B12催化剂,并且也具有广泛的应用(图2, B-D)。天然维生素B12的咕啉环仍然存在于上述配合物种,因此中心钴离子合成B12衍生物的氧化还原潜力和电子属性类似于天然B12,这表明合成B12 衍生物在分子转换中显示出像有机酶一样的高反应性。6)

3. 使用B12配合物的有机电反应
如上所述,B12衍生物的亲核Co(I)与有机卤化物反应,在生成的烷基化合物的同时随后脱卤反应。因此,它很可能会发展成为一种活性Co(I)类形成烷基化配合物的方法,这是一个B12依赖酶反应的关键中间体。合成的B12配合物可以通过化学还原试剂比如硼氢化钠、金属锌、钠汞齐等还原, 因此大量使用这些化学试剂是不符合绿色化学的。因此电化学技术被用于生成B12配合物Co(I)类,在这个反应中,B12配合物作为电反应中介物(图3)。7)
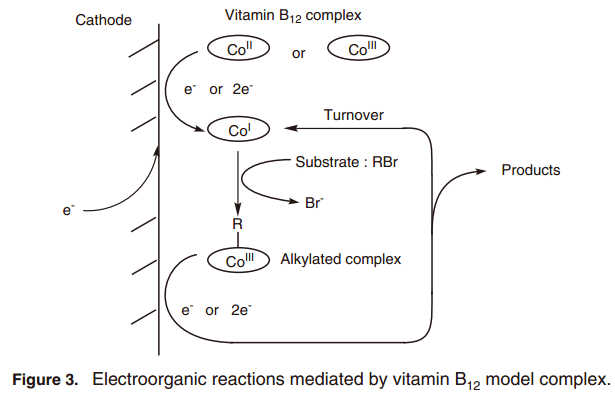
我们通过控制电位电势在-1.4或者-1.5Vvs来开发各种分子变换. Ag/AgCl利用疏水性维生素B12作为一个中介物(方案3)。例如, 当溴代丙烯酸酯作为基质,通过烷基配合物的均裂分子内环化有机自由基合成大圆环内酯(方案3, eq. 1).8)中间体烷基化配合物形成利用电喷雾电离质谱(ESI-MS)和紫外-可见光谱通过检测电解溶液被直接观测到。疏水性维生素B12也能有效地充当环境污染物的脱氯催化剂,例如如二氯二苯基三氯乙烷(DDT)(方案3, eq. 2). 9)值得注意的是,在反应过程中,由于氯原子脱氯处理成无害的氯离子,B12的催化体系不产生有毒二次污染物如氯气和光气(方案2)。此外, B12催化剂反应后不会降解。它的高耐用性在反应过后通过UV-vis和质谱分析后确认。当其他的钴配合物,如卟啉配合物,在相同条件下,电解使用过后复合物严重退化。
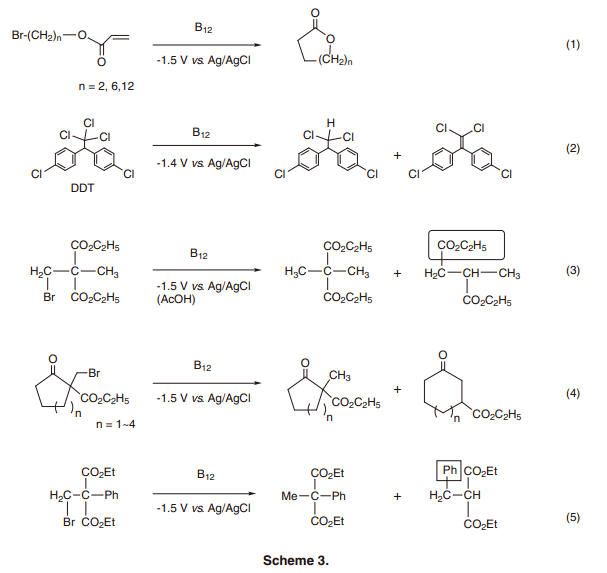
为了重复使用这个强大的B12催化剂, B12配合物被固定在电极上或者溶解在离子溶液中用于试验过后回收或者循环使用。在前者的情况下,B12配合物被固定在铂电极的表面上,通过引入一个三甲氧基硅烷基侧链,覆盖率约在1.6×10–10mol/cm2(2, B)。使用苯乙级溴作为模板的反应性能进行了评估,证明反应活性很高,6,000周转数/h(图4)。10) 反应结束后,产物和B12催化剂可以很容易的进行分离,因为催化剂被固定在电极上面。其他的B12修饰电极也有了报道,比如说聚合物覆盖电极,界面聚合电极,和溶胶-凝胶的FLM掺杂电极。11-13)
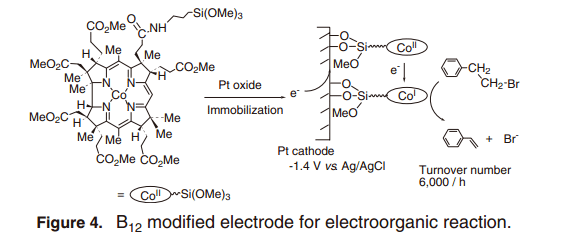
另一方面,当离子液体作为反应溶剂,B12配合物作为离子溶液,显示出以下各种好处。一般情况下,有机电反应在含有电解质的极性有机溶剂中反应。然而,使用含有多种电解质的有机溶剂的这种反应相对于当前节约使用化学试剂的理念是不可取的,即使是该程序被用于降解氯化有机化合物。最近, 离子溶液中有机电反应被越来越多的报道用于解决这些问题。离子液体在通常压力下是室温液体盐,其特点是由它的不易挥发性,防火阻燃,和优异的导电性,并且是优异的有机电反应溶剂。14)离子溶液中使用B12配合物作为催化剂,DDT电解发生,同样的方式,含有支持电解质的极性溶剂中脱氯反应同样的有效的进行。此外,反应结束后的产品和B12配合物的提取在有机层和离子液体层中分别提取。反应中溶于离子液体的B12配合物可以回收再用(图5)。15) 更有趣的是, 离子液体中B12配合物的反应性提高,即在二甲基甲酰胺(DMF)反应中,基板的转换效率被提高了四倍。这种在DMF的离子液体增强的反应可以被解释为休斯英戈尔德预测中的溶剂的极性对反应速率的影响。16) CO(I)与DDT产生的电反应是一种“Menschutkin type of reaction”其中两个中性反应物Co(I) 和DDT, 反应形成的带电产品在极性离子液体中通过电荷分离的活化络合物,这最终降低激活能量,导致反应速率的增加。类似的促进效果反应在对-硝基苯磺酸甲酯与三-n-丁胺离子液体中呗密切观测到。17)其实,离子液体中ETN value18) 已经被确认为极性指数。例如, 1-正丁基-3 -甲基咪唑四氟硼酸盐的指数为0.673, 表明它是高极性的极性溶剂,例如DMF (ETN= 0.386). 19)
4. B12配合物的光敏性反应
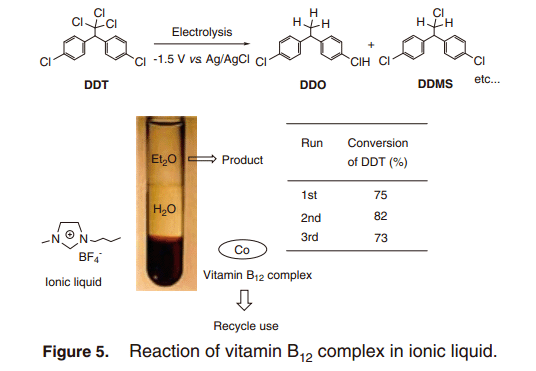
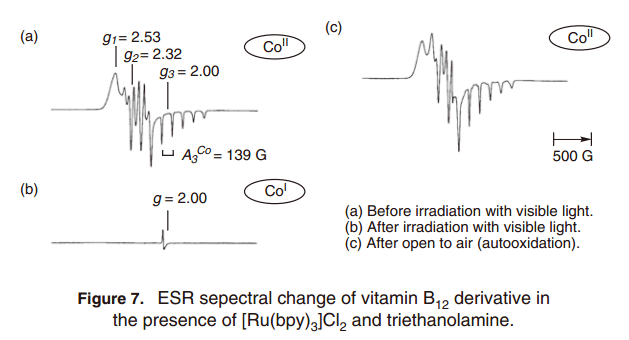
我们试图设计一个光驱动催系统,可以利用B12 配合物开发一个清晰的分子转换。光是地球上丰富,清洁的能源之一,并已用于在有机合成中的很长一段时间。因此,我们注意到了三联吡啶配合物钌(II)([RuII(bpy)3]Cl2), 一直广泛的用作光敏剂,并使用的光诱导电子转移构建了催化系统用于钴(I)类的制造反应。20) [RuII(bpy)3]Cl2配合物在可见光照射下活跃,且[Ru(bpy)3]+ 配合物具有很高的还原电势(–1.35 V vs. Ag/AgCl) ,通过牺牲电子给体三乙醇胺还原淬火得到。21) 因此,它能够由从钌光敏剂的电子转移产生催化活性B12 配合物(E1/2(CoII/CoI) =–0.6Vvs. Ag/AgCl)。如图6所示, 在钌光敏剂和牺牲给电子供体参与下,利用B12配合物DDT的脱氯反应发生。因此, 三小时内大部分DDT 被转换为DDD, 一个单脱氯产品。在没有B12配合物或者是黑暗条件下反应几乎没有进展。因此,人们认为Co(I)是通过光诱导电子转换反应产生,可以作为引发反应的活性体。ESR谱更改证实,钌光敏剂电子转移反应还原了B12配合物生成钴(I)的物种。ESR特征信号用于检测B12顺磁性Co(II),而相应的ESR信号用于光反应中反磁性Co(I)(图7).
5. B12-TiO2混合催化剂
最近,由金属络合物和光敏材料半导体组成的混合催化剂已经有所报道。22-23)根据该技术,很有可能开发出一种金属络合物与半导体互为效果的混合催化剂。因此,我们专注于二氧化钛,24)这是一个n-type半导体,用于开发一个光源驱动B12-TiO2混合催化剂。氧化钛(TiO2)-传导带电子能量的降低(–0.5 V vs.正常氢电极(NHE)) 25) 使B12 络合物还原成具有催化活性的Co(I) species, 这表明光能驱动混合催化剂可以通过固定B12络合物于TiO2上开发合成。在这种情况下,二氧化钛不仅起到了B12络合物的脚手架的作用,但还担任还原B12络合物的电子源(图8)。这里,通过多个维生素B12的羧基(图2,C型)和TiO2表面羟基基团的相互作用将B12络合物固定在TiO2上。约3-4×10-5摩尔(40-50毫克)的维生素B12络合物(cobyrinic酸)被发现固定在1克二氧化钛上面,70%-80%的TiO2表面覆盖着B12络合物。B12络合物被牢固打的固定在TiO2表面上,保持稳定1年以上,甚至后期分散于各种有机溶剂,如醇,并不通过超声波处理分离。

当氯化有机化合物比如说DDT的降解反应, 利用此混合催化剂进行反应时,仅仅几毫克的催化剂便可在一天时间里激活DDT使得脱氯反应100倍作用。26)混合催化剂通过光能激活,因此化学试剂或者昂贵的反应器都不需要。不可见光(365 nm) 所提供的紫外线照射为激发TiO2提供足够的能量。此外, B12络合物和TiO2都是无毒的,因此B12-TiO2混合催化剂可能是环境/人类友好型催化剂。另外, 我们研究有机合成中使用混合催化剂的自由基反应,该结果表明这种混合催化剂可应用于各种分子转换,包括上述环扩反应(方案3,式4)和1,2-迁移的官能团(方案3,式5)。因此,混合催化剂作为一种替代,可以使用tin化合物(Bu3SnH/AIBN system)用于常规的自由基合成试剂。
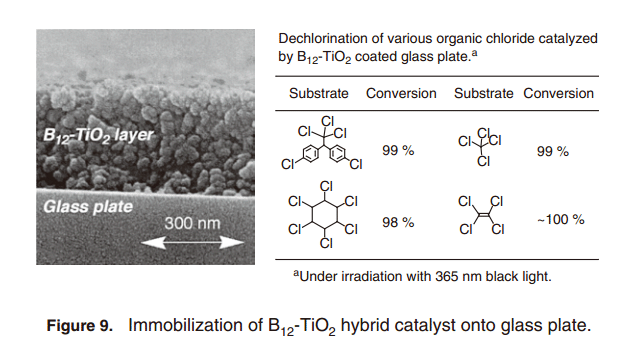
当它被转换成浆料溶胶溶液的时候,二氧化钛也可以被固定在载体上,如玻璃基板和珠子上面。TiO2薄膜(约几百纳米厚度), 通过在玻璃基质上浸涂得到,通过浸入到含羧基集团的B12络合物溶液,与B12 络合物进行杂交反应。因此,在玻璃板上面制备B12-TiO2混合催化剂也可以催化上述反应(图9)。玻璃板面上固定B12-TiO2混合催化剂可以简化反应过程,并且,以便于从反应混合物中分离产物。
6. 维生素B12-超支化聚合物复合催化剂
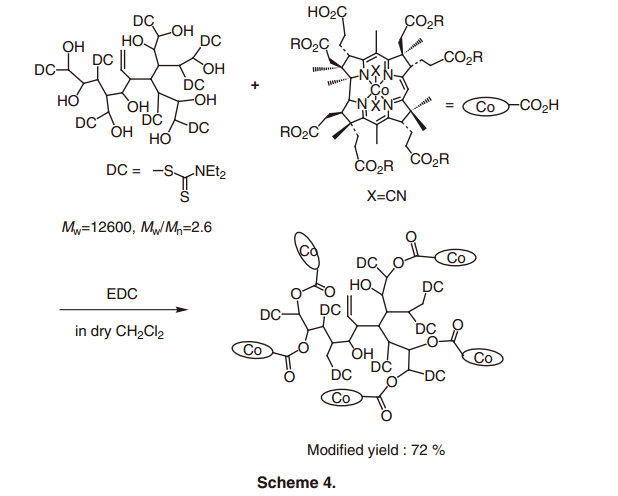
超支化聚合物是树突状聚合物打的一种,通过简单经济的方法单步骤合成,与通过多步聚合反应合成的树枝状聚合物相比,它具有各种优点。27)此外,与线性聚合物相比,该聚合物具有较高的溶解性和较低的溶液粘度。它预期可以使用在一个均匀的溶液中。28)事实上, 超支化聚合物,由于其高度支化结构有许多末端官能团,适合携带官能分子。29)考虑到由超支化聚合物提供的纳米空间作为一个新的反应领域,B12-超支化聚合物复合催化剂得以合成(方案4)。30) 所使用的维生素B12配合物的量可以在超支化聚合物的末端官能团调整到某处之间的几%和70%,并且可以容易地控制密度的B12配合物。当B12配合物密度确定,相邻的复合物的合作效果便于苯乙基溴的二聚化反应。31)
7. 结语
我们概述了从B12依赖酶到分子转换的发展过程。混合性催化酶包含金属配合物的合成,与B12酶的活性中心相似,激活各种分子变换,包括环境友好的有机合成反应和有机卤化物污染物的降解反应的有机电反应或光化学反应。我们推出的这种仿生化学的发展将对下一代的科学和技术起到重要的作用。
参考文献
1.(a) Vitamin B12 and B12-Proteins ed. by B. Kräutler, D. Arigoni, B. T. Golding, Wiley-VCH 1998; (b) Chemistry and Biochemistry of B12 ed. by R. Banerjee, Wiley-Interscience 1999.
2.(a) D. C. Hodgkin, J. Pickworth, J. H. Robertson, K. N. Trueblood, R. J. Prosen, J. G. White, Nature 1955, 176, 325-328; (b) D. C. Hodgkin, J. Kamper, M. Mackay, J. Pickworth, K. N. Trueblood, J. G. White, Nature 1956, 178, 64-66; (c) P. G. Lenhert and D. C. Hodgkin, Nature 1961, 192, 937-938.
3.(a) R. Banerjee and S. W. Ragsdale, Annu. Rev. Biochem. 2003, 72, 209-247; (b) T. Toraya, Chem. Rev. 2003, 103, 2095-2127; (c) B. Kräutler, Biochem. Soc. Trans. 2005, 33, 806-810.
4.(a) G. Glod, U. Brodmann, W. Angst, C. Holliger, R. P. Schwarzenbach, Environ. Sci. Technol. 1997, 31, 3154-3160; (b) C. Holliger, G. Wohlfarth, G. Diekert, FEMS Microbiol. Rev. 1999, 22, 383-398; (c) B. Kräutler, W. Fieber, S. Ostermann, M. Fasching, K. –H. Ongania, K. Gruber, C. Kratky, C. Mikl, A. Siebert, G. Diekert, Helv. Chim. Acta 2003, 86, 3698-3716; (d) S. Ruppe, A. Neumann, G. Diekert, W. Vetter, Environ. Sci. Technol. 2004, 38, 3063-3067; (e) K. M. McCauley, D. A. Pratt, S. R. Wilson, J. Shey, T. J. Burkey, W. A. van der Donk, J. Am. Chem. Soc. 2005, 127, 1126-1136; (f) J. E. Argüello, C. Costentin, S. Griveau, J. –M. Sáveant, J. Am. Chem. Soc. 2005, 127, 5049-5055.
5.Reviews: (a) Y. Hisaeda, T. Nishioka, Y. Inoue, K. Asada, T. Hayashi, Coord. Chem. Rev. 2000, 198, 21-25; (b) K. L. Brown, Chem. Rev. 2005, 105, 2075-2150.
6.(a) Y. Murakami, Y. Hisaeda, A. Kajihara, Bull. Chem. Soc. Jpn. 1983, 56, 3642-3646; (b) Y. Murakami, Y. Hisaeda, A. Kajihara, T. Ohno, Bull. Chem. Soc. Jpn. 1984, 57, 405-411.
7.R. Scheffold, G. Rytz, L. Walder, Modern Synthetic Methods, Vol. 3, ed. By R. Scheffold, John Wiley & Sons, 1983, 355-440.
8.H. Shimakoshi, A. Nakazato, T. Hayashi, Y. Tachi, Y. Naruta, Y. Hisaeda, J. Electroanal. Chem. 2001, 507, 170-176.
9.(a) H. Shimakoshi , M. Tokunaga, Y. Hisaeda, Dalton Trans. 2004, 878-882; (b) H. Shimakoshi, Y. Maeyama, T. Kaieda, T. Matsuo, E. Matsui, Y. Naruta, Y. Hisaeda, Bull. Chem. Soc. Jpn. 2005, 78, 859-863.
10.H. Shimakoshi , M. Tokunaga, K. Kuroiwa, K. Kimizuka, Y. Hisaeda, Chem. Commun. 2004, 50-51.
11.(a) A Ruhe, L. Walder, R. Scheffold, Helv. Chim. Acta 1985, 68, 1301-1311; (b) Y. Murakami, Y. Hisaeda, T. Ozaki, Y. Matsuda, J. Chem. Soc., Chem. Commun. 1989, 1094-1096; (c) D.-L. Zhou, C. K. Njue, J. F. Rusling, J. Am. Chem. Soc. 1999, 121, 2909-2914; (d) C. K. Njue and J. F. Rusling, Electrochem. Commun. 2002, 4, 340-343.
12.R. Fraga, J. P. Correia, R. Keese, L. M. Abrantes, Electrochim. Acta 2005, 50, 1653-1655.
13.H. Shimakoshi, A. Nakazato, M. Tokunaga, K. Katagiri, K. Ariga, J. Kikuchi, Y. Hisaeda, Dalton Trans. 2003, 2308-2312.
14.T. Welton, Chem. Rev. 1999, 99, 2071-2083.
15.M. Jabbar, H. Shimakoshi, Y. Hisaeda, Chem. Commun. 2007, 1653-1654.
16.K. A. Cooper, M. L. Dhar, E. D. Hughes, C. K. Ingold, B. J. MacNulty, L. I. Woolf, J. Chem. Soc. 1948, 2043-2046.
17.L. Crowhurst, N. L. Lancaster, J. M. P. Arlandis, T. Welton, J. Am. Chem. Soc. 2004, 126, 11549-11555.
18.C. Reichardt, Solvent and Solvent Effects in Organic Chemistry, VCH (UK) Ltd., Cambridge, UK, 2nd edn, 1998.
19.(a) S. N. V. K. Aki, J. F. Brennecke, A. Samanta, Chem. Commun. 2001, 413-414; (b) M. J. Muldoon, C. M. Gordon, I. R. Dunkin, J. Chem. Soc., Perkin Trans. 2, 2001, 433-435.
20.(a) H. Shimakoshi , M. Tokunaga, T. Baba, Y. Hisaeda, Chem. Commun. 2004, 1806-1807; (b) H. Shimakoshi, S. Kudo, Y. Hisaeda, Chem. Lett. 2005, 34, 1806-1807.
21.A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser, A. V. Zelewsky, Coord. Chem. Rev. 1988, 84, 85-277.
22.(a) S. O. Obare, T. Ito, G. J. Meyer, Environ. Sci. Technol. 2005, 39, 6266-6272; (b) Y. Astuti, E. Palomarse, S. A. Haque, J. R. Durrant, J. Am. Chem. Soc. 2005, 127, 15120-15126; (c) S. O. Obare, T. Ito, G. J. Meyer, J. Am. Chem. Soc. 2006, 128, 712-713; (d) B. I. Ipe and C. M. Niemeyer, Angew. Chem. Int. Ed. Engl. 2006, 45, 504-507.
23. R. J. Kuhler, G. A. Santo, T. R. Caudi l l, E. A. Betterton, R. G. Arnold, Environ. Sci. Technol. 1993, 27, 2104-2111.
24.(a) M. A. Fox, Acc. Chem. Res. 1983, 16, 314-321; (b) M. D. Ward, J. R. White, A. J. Bard, J. Am. Chem. Soc. 1983, 105, 27-31; (c) M. A. Fox and M. T. Dulay, Chem. Rev. 1993, 93, 341-357; (d) M. R. Hoffmann, S. T. Martin, W. Choi, D. W. Bahnemann, Chem. Rev. 1995, 95, 69-96.
25.A. Fujishima, T. N. Rao, D. A. Tryk, J. Photochem. Photobiol. C: Photochem. Rev. 2000, 1, 1-21.
26.H. Shimakoshi, E. Sakumori, K. Kaneko, Y. Hisaeda, Chem. Lett. 2009, 38, 468-469.
27.M. Jikei and M. Kakimoto, Prog. Polym. Sci. 2001, 26, 1233-1285.
28.P. H. Toy and K. D. Janda, Acc. Chem. Res. 2000, 33, 546-554.
29.(a) K. Ishizu and A. Mori, Polym. Int. 2001, 50, 906-910; (b) K. Ishizu, T. Shibuya, A. Mori, Polym. Int. 2002, 51, 424-428; (c) K. Ishizu, T. Shibuya, J. Park, S. Uchida, Polym. Int. 2004, 53, 259-265.
30.K. Tahara, H. Shimakoshi, A. Tanaka, Y. Hisaeda, Tetrahedron Lett. 2007, 48, 5065-5068.
31.K. Tahara, H. Shimakoshi, A. Tanaka, Y. Hisaeda, unpublished result.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号