三氟乙酰亚氨卤化物作为常用的氟化物衍生物
- 2012-12-11
- 专题
一、三氟亚氨基卤化物介绍
含有三氟甲基基团的有机化合物在医药、农药和电子材料如液体晶体的关键功能中扮演重要角色。以下介绍几种三氟甲基(-CF3)转化为有机化合物的三种方法:1,利用含(-CF3)的化合物;2,三氟甲基衍生物使用三氟甲基合成制剂如化合物CF3-TMS、FSO2CO2Me、CF3等。3,利用氟化剂如F2、HF将CCl3和COOH基团转化为-CF3。方法3是大规模工业生产合成含有-CF3化合物的传统方法,合成的分子大部分是结构简单、性质稳定的分子。另一方面,方法1和2在实验室已经用于结构复杂和具有价值的化合物的合成。1)
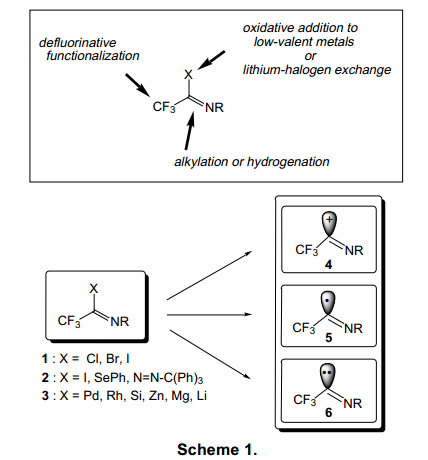
现今没有太多的含有-CF3化合物应用于商业生产,因此开发合成复杂结构的含有-CF3化合物非常重要。他们应该在高收率的前提下,从容易合成的、含有高度潜在功能的分子着手,再作进一步修饰分子。在这个基础上,根据以下前沿文献显示,三氟亚氨基卤化物是一个独特的有价值的含-CF3化合物:a:可以一步高收率的由三氟乙酸合成,b:可以相对稳定的贮存 c:含有高度潜能的基团如CF3、亚氨基的C=N双键和卤素(图1)。
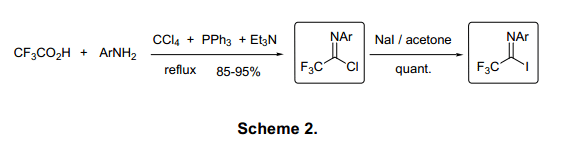
(综述):利用三氟乙酸在高收率(85%-95%)的前提下合成亚氨基卤化物(X:Br、Cl)的方法(图2)。2)由于四氯化碳(CCl4)禁止使用,还可以用PPh3Cl2代替四氯化碳。在工业生产制造中,相应的可以在磷酰氯的作用下由三氟乙酰胺转化成亚氨基氯。2c)
亚氨基碘1(X:I)的合成是相应的氯化物、NaI按一定比例加入丙酮中反应,使分子中的Cl交换为I。由于亚氨基氯相对比较稳定,因此可将未反应的亚氨基氯通过硅胶柱层析法分离出来循环使用。三氟亚氨基卤化物在酸性或中性条件下可以缓慢水解,但在基本条件下反应迅速。相反不含氟的亚氨基卤化物一般迅速与水反应形成酰胺。稳定的酸性源自亚氨基受-CF3吸电子的影响自身的质子化。
(反应):亚氨基卤化物通过碳正离子4、自由基5、碳负离子6(图1)形式广泛应用于有机合成。例如,氯化物1(X=Cl)可与亲核物质发生亲核取代反应或在酸性催化作用下进行傅克反应,将氯转换到其他官能团上。碘基、硒基、偶氮亚氨基化合物2通过光催化反应或热化学反应生成碳自由基5,它可以生成烯烃、炔烃、芳香烃化合物。亚氨基卤化物1可以通过氧化加成形成低价过渡金属化合物或者卤素金属交换反应转化成相应的亚氨基金属化合物3,也可以与亲电物质发生亲电反应或过度金属催化交叉耦合反应(图1)。
2 三氟乙酰亚氨基卤化物与亲核试剂的反应
2.1含氧亲核试剂
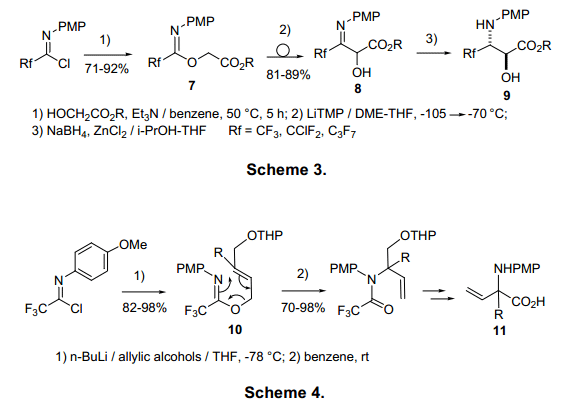
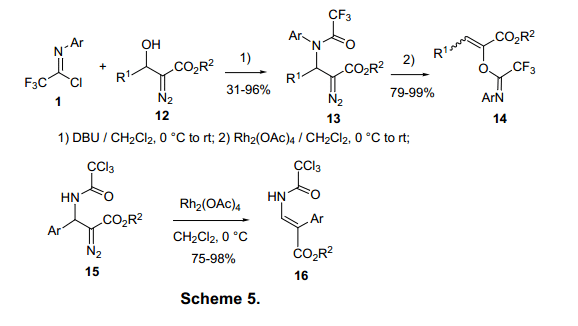
由于亚氨乙酰卤的亚氨基碳有强亲电性,它在碱催化温和条件下易于醇发生反应生成相应的亚氨酸酯7和10,该反应收率较高。(图3,4)每个亚氨酸酯73)和104)可通过重排反应合成氨基酸衍生物。重排动力来自具有比亚氨酸酯7更高热力学稳定性的相应的酰胺。亚氨基氯化物1与重氮醇化物12反应生成酰胺13(图5)。5)由13生成的碳烯中间体被分子内的氨基羰基氧进攻,紧接着重排形成14.该重排高度依赖于取代基。例如,三氯乙酰胺通过1,2—芳环转换形成16.在整个重排过程中12芳环上的C-3通过imidate-amide重排成为14的C-2。
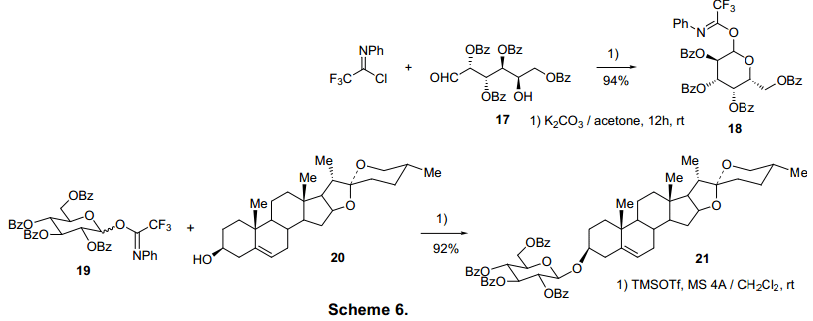
自从yu等人报道19与醇反应具有高反应活性并且在路易斯酸作用6b)下有较好的离去能力,19常常被用来糖基化反应。6)三氟乙酰亚胺酯(糖基供体)与乙醇(糖基受体)可以顺利的在温和的条件下反应,具有较高收率如表6。
2.2含氮亲核试剂
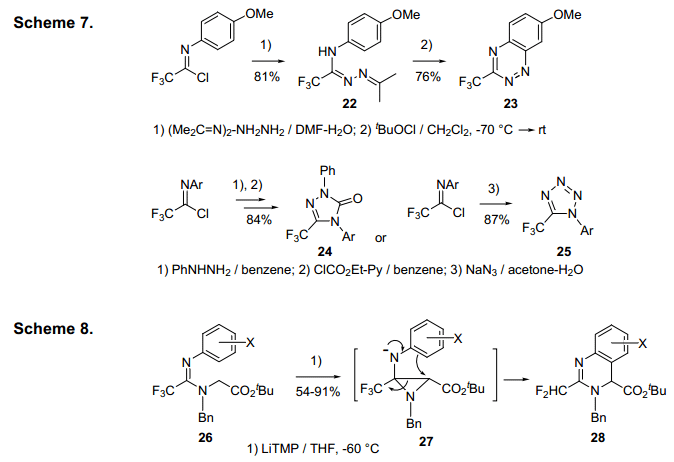
亚氨基氯化物1中的氯可以通过含氮的亲核试剂平稳地取代给各种氨基酰胺 ,这可以被转换为有用的三氟甲基含氮杂环化合物。一些合成应用化合物如氧化环化化合物22与次氯酸叔丁酯(t-BuOCl)作用生成CF3-benzotriazine 23,7)与苯肼反应跟着通过缩合环化形成三氟甲基三唑化合物24,通过环化作用形成亚氨基 叠氮化物即三氟甲基三唑化合物258)(图7)。含二氟甲基 的间二氮杂萘28的合成是通过连续的环化脱氟作用及连续通过氮杂环丙烷中间27,而27是从imidamide 26合成而来的(图8)。9)
2.3.与碳亲核试剂的反应
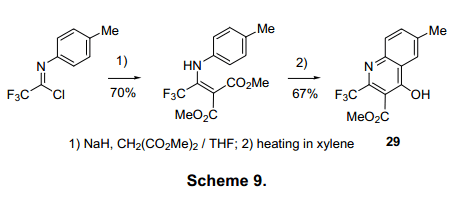
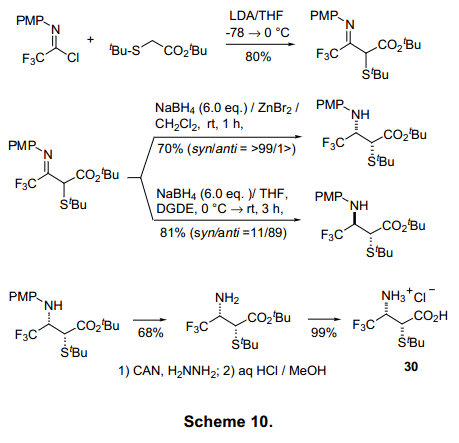
亚氨基氯化物1还可以与碳亲核试剂会发生反应,因此它通常用于含有-CF3化合物的合成。合成的例子如2-CF3-取代喹诺酮类2910)和具有非对映体异构的2-S-3-氨基丁酯3011)的合成分别列在图表9和图表10。
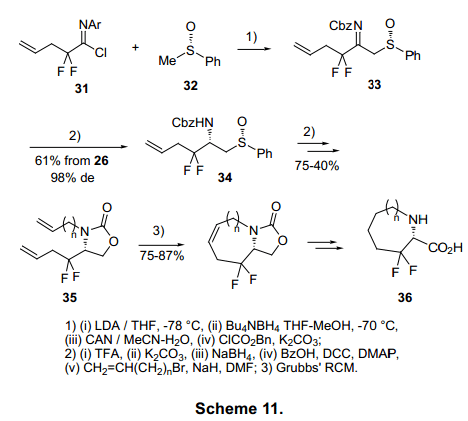
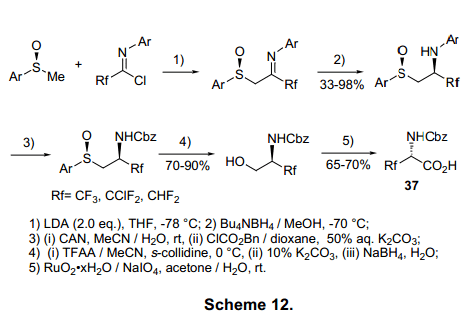
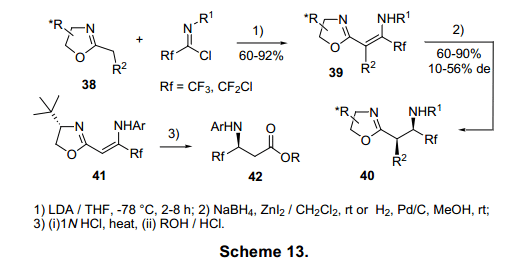
Fustero等人已经通过双氟亚氨基氯化物31和光学活性亚砜32合成了光学纯环氨基酸36,紧随其后的是非对应异构体减少亚氨基,用亚硫酰基作为手性辅助物。闭环化合物在Grubbs催化下通过复分解反应衍生出b,b-二氟环状氨基酸36(图11)。12)与5号位和6号位有关的b,b-二氟环状氨基酸已由相关的亚氨基氟化物合成。13)同样的方法已用于合成三氟、二氟、氯代二氟丙氨酸37(图12)的合成。14)减少非对应异构体亚烯胺38可与亚氨基氯化物合成手性恶唑啉39,并进一步合成b-氨基酸衍生物40。此法可以合成光化学纯b-氨基酸衍生物42(图13)。15)
2.4 分子内碳的亲核反应
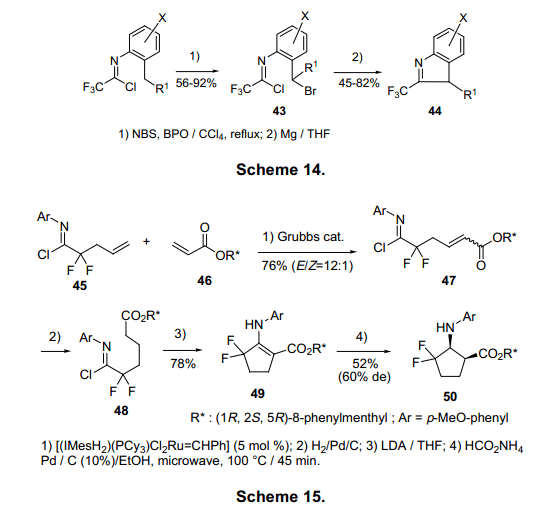
亚氨基分子内碳的亲核反应已用于含氮杂环化合物的合成。亚氨基氯化物43的苄基位溴可以有选择性的与镁反应,即使有芳环上的碳-氯成2-CF3-吲哚衍生物44(图14)。16)氢化后47的紧接着分子内烃化生成b-氨基-a,b不饱和环酯49,分子47是由二氟亚氨基氯化物45与手性丙烯酸酯46反应合成。非对应异构体49加氢生成五环氨基酸50。(图15)17)
3 三氟亚氨基卤化物及衍生物的自由基反应
当三氟亚氨基卤化物X是碘、硒、偶氮当三氟乙酰亚胺衍生物2的官能团有碘基、硒基或含氮基团,在光照或加热条件下就会产生相应的激进物质5,这就会触发它与烯烃、炔烃的激进反应。图表16展示了这两种分子内的和分子间的反应机理。化合物51与苯乙炔在光反应条件下产生的互为同分异构体52和53的混合物。攻击由乙烯基激进的中间体的ipso位置和斯皮罗环C-N键的断裂会导致1,2位的迁移从而生成化合物52。而另一方面,打破的C-C键的1、2位的迁移会导致另一个化合物53的形成。18b)在光照条件下化合物54会生成含有3-Ketoindole的化合物55.这个分子内的C-I键形成C-C三键,通过由imidoyl激进形成的中间和一部分后来水解反应形成的iodoalkylidene作用形成一个酰基从而生成含有3-Ketoindole的化合物55,作为最终产物。18a)
4 三氟乙酰亚胺卤化物与金属化合物的反应
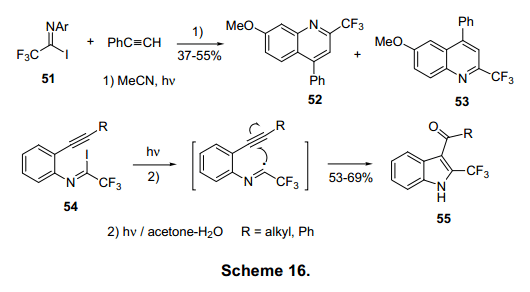
三氟乙酰亚胺金属化合物57可能适用各种各样的通过金属碳碳键的形成三氟甲基氮化合物的合成。三个活泼的基团56,57,和58都可适用于作为X基团如图表17.许多报道已经发表了尤其是关于异丙烯基金属化合物56的报告。19)另一方面,三氟乙酰基金属化合物58非常不稳定的,只有三氟乙酰基钯类化合物被用于有机合成。20)我们团队已经广泛研究了三氟亚氨代乙酰基金属化合物57。三氟亚氨代乙酰基金属化合物57化学结构,它的合成方法,化合物的性能,有关的化学反应以及它的应用已经被描述在我们的文章中。
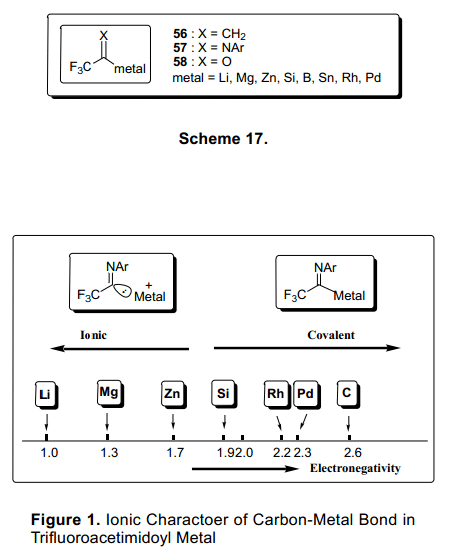
三氟亚氨代乙酰基金属化合物57的稳定性主要来源于碳金属共价键的稳定性。碳原子与金属原子之间很小的电负性差异就能提供很高的共价键稳定性(图1)。例如,含Pd金属化合物在130°C条件下还能在很长一段时间内具有很好的稳定性。与此同时,有机锂化合物必须保存在一个至少低于-60°C的条件下,尽管有机锂化合物在大多数反应中充当碳负离子。亚氨基有机锂化合物59的形成是由丁基锂通过方法1与碘在原位交换,进而迅速与亲和物质反应形成60。在-60°C以上时,不稳定的锂化物59形成中间体60产生二聚体。(图18)21)相应的Zn化物与Zn—Al在DMF中保存比Li化物稳定,并且可与亲电物质反应。(图19)22)
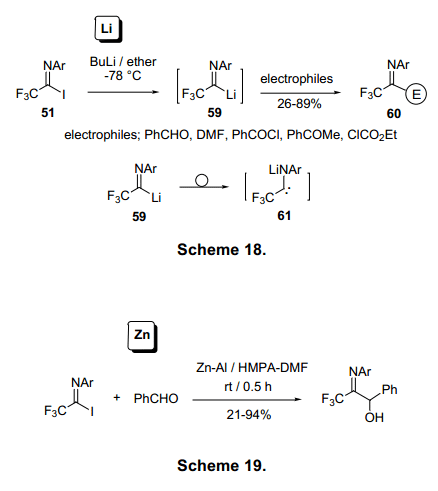
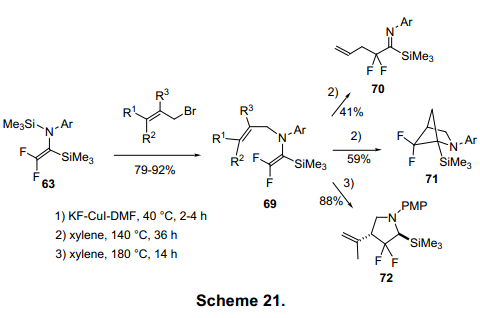
亚氨基镁由亚氨基氯与保存在TMS-Cl中的Mg在TFH中反应制得。Mg基化合物在0°C保存相对较稳定。具有选择性的甲硅烷基化1的亚氨基碳在-700°C生成亚氨基硅烷62,产率约为70%。(图20)23)有趣的是,该反应在0°C时高产率的生成双甲基硅烷63。连续的Mg催化使C-Cl和C-F激活,形成63。这个双甲基硅烷烯胺63有三个反应点:N、C-1、C-2。逐步激活的N和C-1与KF反应生成64。同时,激活的N在路易斯酸作用下,C-1与KF反应生成65。63在路易斯酸催化作用下C-2与苯甲醛烷基化形成65a。然后,苯甲酸化合物66b被转换成4-羟基-3,3-二氟-2-丁基氨基酸的前体68b(图20,21)。24)63在F离子催化剂作用下脱去甲硅烷基,并在氨基N上进行烯丙基化生成69,69经过aza—Claisen重排生成二氟化合物70,71,72图21)。25)
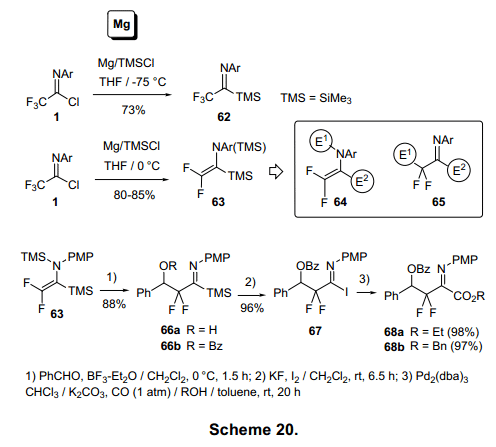
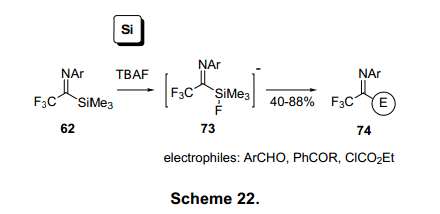
在F离子催化下脱甲硅烷基的62可以生成五价硅酸盐中间体73,等量亚氨基的碳负离子比锂化物更稳定,并且更已保存,它可以在50°C进行烷基化为74(图22)。26)因此,亚氨基硅烷-62可以用于亲电性较弱的物质反应。
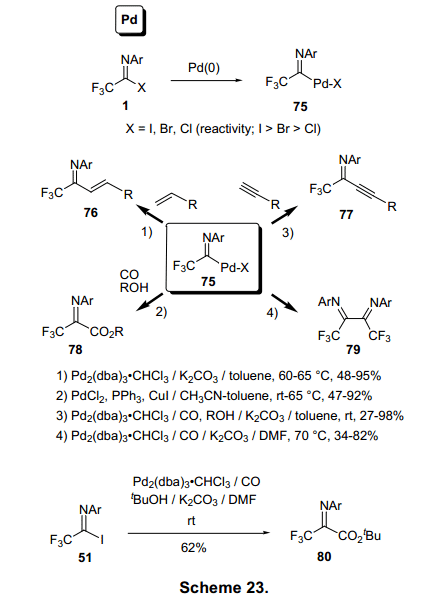
含Pd化合物75可以由任何亚氨基卤化物(X:I、Br、Cl)制得。然而,低化合价的钯决定卤化物的氧化加成反应速率,因此,碘代亚氨基卤化物最有用的反应是氧化加成和亚氨基卤化物与亲核试剂的亲核取代的反应,两种反应相互竞争,尤其是与亲核试剂的反应超过了氧化加成反应的反应速率。钯金属亚氨基卤化物75被用来生成各种碳碳键和如图表23和24所示的化学反应。与1-烯烃反应的Heck-Mizorogi反应和与炔烃反应的Sonogashira反应分别被非常成功地用来制备ene-imines 和 yne-imines。27)Pd催化作用下,1(X=I)被烷氧基化,它与伯醇如含有苄基和苯基生成收率较高的3,3,3,-三氟-2-亚氨基丙酸酯,该物质是合成三氟乙酸酯类物的原料。28)值得注意的是甚至3-丁酯类化合物78(R=t-Bu)可以在DMF或DMI作溶剂中以60-70%的收率制得。29)在缺少亲和物质的情况下,75的二聚体可通过Pa类物质合成α-二亚胺类化合物79。(图23)30)
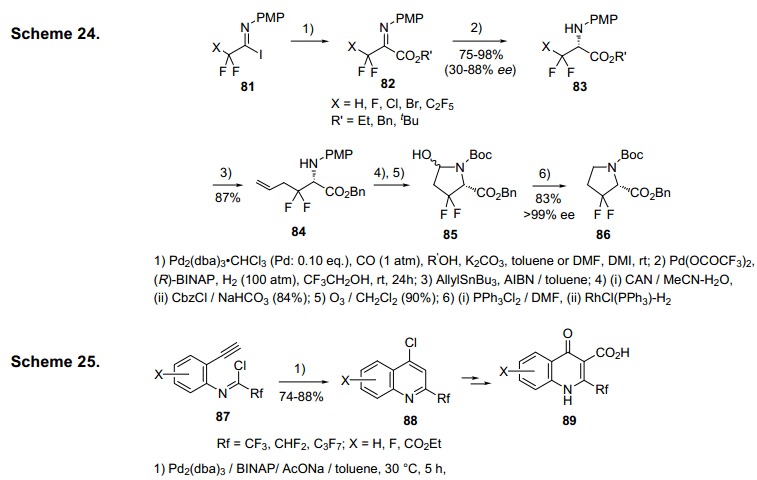
溴基二氟乙酰亚氨基碘化物81可由b,b-二氟脯氨酸86合成(图24)。31)酯82是从亚氨基碘代物81在Pd催化被烷氧基化(carboalkoxylation)及其它的亚氨基的基团在Pd (OCOCF3)2催化剂条件下受到下不对称氢化作用,在三氟乙醇32)的条件下生成化合物83,产率为88%。化合物83的烯丙基自由基和84强化的对映体通过再结晶紧随其后臭氧化、脱水,然后化合物85经氢化作用,经光化作用合成纯的脯氨酸化合物86。31)
含有一个卤素原子的亚氨基卤化物1迄今为止还没有通关实验合成验证的(图23)。然而,合成含有一个亚氨基和一个卤素原子的产品会增加其附加价值。图表25展示了一个例子:含有一个卤素原子和一个亚氨基的化合物可以被有效的利用在化学合成中。33)在4位含有一个氯原子的喹啉环可以有效地用来合成喹诺酮羧酸89。
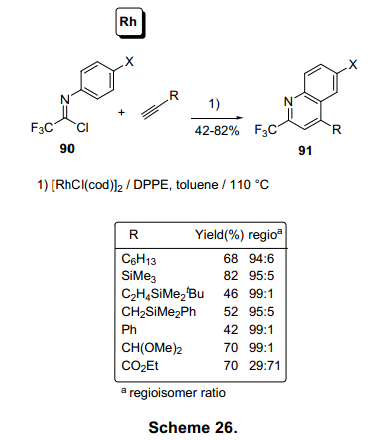
铑催化剂也可以激活亚氨基氯化物90。在化合物90的N-芳基环的邻位引入炔烃,亚氨基碳通过亚氨基和铑催化剂共同作用生成喹啉环91。与各种含炔基化合物反应可以有效地代替喹啉骨架(图26)。34)
5结论
如今,约有百分之十的现有的商业化药物含有氟原子或具有氟功能,这点明显增强了他们的生物活性。因此,氟化物被人们广泛的关注和利用。然而,合成有机氟化学最大的问题之一寻找是一个较小的对于目标分子具有可用性的起始氟化合物。在此基础上,有机合成化学家正负责开发多才多艺有机氟合成块,此合成块可以通过传统的反应提供高度可用的起始基物如三氟乙酸。三氟亚氨基乙酰化卤代物(Trifluoroacetimidoyl)是非常有用和可靠的化合物,这正是有机氟化学所需求的合成产品。他们正在准备开发这样的一个反应,在非常传统的条件三氟乙酸只通过一个步骤的反应就能得到目标产物,并且具有很好的产量。这个亚氨基卤化物1为我们提供了通用的具有反应活性的亚氨基碳正离子,自由基和碳负离子,特别是亚氨基金属物(imidoyl metals),所有这一切都是可靠的有机合成方法并且将来会被更加广泛地利用。
参考文献
1) a) Japan Society for Promotion of Science, 155 Fluorine Chemistry Committee Ed. “Introduction to
Fluorine Chemistry”, 2004, Sankyo Publishing Co. Ltd,Japan; b) Uneyama, K. “Organofluorine Chemistry”,2006, Blackwell Publishing, Oxford, UK
2) a) Tamura, K.; Mizukami, H.; Maeda, K.; Watanabe,H.; Uneyama, K. J. Org. Chem. 1993, 58, 32-35;
b) Synthesis of imidoyl chloride 1 by the radicalreaction of isonitriles with CF3I: Huang, W. S.; Yuan,C. Y.; Wang, Z. Q. J. Fluorine Chem. 1995, 74, 279-282; c) Industrial scale production of 1, Hagiya, K.;
Sato, Y.; Koguro, K.; Mitsui S. PCT Int. Appl. WO2005-035484.
3) Uneyama, K.; Hao J.; Amii, H. Tetrahedron Lett. 1998,39, 4079-4082.
4) Berkowitz, D. B.; Wu, B.; Li, H. Org. Lett. 2006, 8, 971-974.
5) Xu, F.; Zhang, S.; Wu, X.-G.; Liu, Y.; Shi, W.; Wang, J.Org. Lett. 2006, 8, 3207-3210.
6) a) Peng, W.; Han, X.; Yu, B. Synthesis 2004, 1641-1647.; b) Yu, B.; Tao, H. J. Org. Chem. 2002, 67,,9099-9102; c) Yu, B.; Tao, H. Tetrahedron Lett. 2001, 42,2405-2407; d) Al-Mahari, N.; Botting, N. P.Tetrahedron Lett. 2006, 47, 8703-8706; e) Bedini, E.;Carabellese, A.; Barone, G.; Parrilli, M. J. Org. Chem.2005, 70, 8064-8070; f) Hanashima, S.; Castagner, B.;Esposito, D.; Nokami, T.; Seeberger, P. H. Org. Lett.2007, 9, 1777-1780; g) Thomas, M.; Gesson, J.-P.;Papot, S. J. Org. Chem. 2007, 72, 4262-4264.
7) Uneyama, K.; Sugimoto, K. J. Org. Chem. 1992, 57,6015-6019.
8) Uneyama, K.; Yamashita, F.; Sugimoto, K.; Morimoto,O. Tetrahedron Lett. 1990, 31, 2717-2718.
9) Hao, J.; Ohkura, H.; Amii, H.; Uneyama, K. Chem.Commun. 2000, 1883-1884.
10) Uneyama, K.; Morimoto, O.; Yamashita, F. TetrahedronLett. 1989, 30, 4821-4824.
11) Ohkura, H.; Handa, M.; Katagiri, T.; Uneyama, K.J. Org. Chem. 2000, 67, 2692-2695.
12) Fustero, S.; Sanz-Cervera, J. F., del Pozo, C.; Acena,J. L. ACS Symposium Series 949 (Current Fluoroorganic Chemistry), 2006, 54-68.
13) six membered compounds: Fustero, S.; Sanchez-Rosello, M.; Rodrigo, V.; del Pozo, C.; Zanz-Cerver, J.F.; Simon, A. Org. Lett. 2006, 8, 4129-4132:five membered compounds: Fustero, S.; Sanchez-Rosello, M.; Sanz-Cervera, J. F.; Acena, J. L.; sel Pozo,C.; Fernandez, B.; Bartolome, A.; Asensio, A. Org. Lett.
2006, 8, 4633-4636.
14) Fustero, S.; Navorro, A.; Pina, B.; Soler, J. G.;Bartolomé, A.; Asensio, A.; Simón, A.; Bravo, P.; Fronza,G.; Volonterio, A.; Zanda, M. Org. Lett. 2001, 3, 2621-2624.
15) Fustero, S.; Salavert, E., Pina, B.; de Arellano, C. R.;Asensio, A. Tetrahedron 2001, 57, 6475-6486.
16) Wang, Z.; Ge, F.; Wan, W. H.; Jiang, J. Hao,J. Fluorine Chem. 2007, 128, 1143-1152.
17) Fustero, S.; Sanchez-Rosello, M.; Sanz-Cervera, J. F.;Acena, J. L.; sel Pozo, C.; Fernandez, B.; Bartolome,A.; Asensio, A. Org. Lett. 2006, 8, 4633-4636.
18) a) Ueda, Y.; Watanabe, H.; Uemura, J.; Uneyama, K.Tetrahedron Lett. 1993, 34, 7933-7934; b) Dan-oh, Y.;Matta, H.; Uemura, J.; Watanabe, H.; Uneyama, K.Bull. Chem. Soc. Jpn. 1995, 68, 1497-1507.
19) Uneyama, K.; Katagiri, T.; Amii, H. Acc. Chem. Res.,submitted for publication.
20) a) Kakino, R.; Shimizu, I.; Yamamoto, A. Bull. Chem.Soc. Jpn. 2001, 74, 371-376; b) Kakino, R.; Yasumi,S.; Shimizu, I.; Yamamoto, A. Bull. Chem. Soc. Jpn.2002, 75, 137-148.
21) a) Watanabe, H.; Yamashita, F.; Uneyama, K.Tetrahedron Lett. 1993, 34, 1941-1944; b) Watanabe,
H.; Yan, F.-Y.; Sakai, T.; Uneyama, K. J. Org. Chem.1994, 59, 758-761.
22) Tamura, K.; Yan, F.-Y.; Takashi, T.; Uneyama, K.Bull. Chem. Soc. Jpn. 1994, 67, 300-303.
23) Akamatsu, C.; Yamauchi, Y.; Kobayashi, T.; Ozeki, Y.;Takagi, J.; Amii, H.; Uneyama, K. Synthesis 2006, 1836-1840.
24) Kobayashi, T.; Nakagawa, T.; Amii, H.; Uneyama, K.Org. Lett. 2003, 5, 4297-4300.
25) Amii, H.; Ichihara, Y.; Nakagawa, T.; Kobayashi, T.;Uneyama, K. Chem. Commun. 2003, 2902-2903.
26) Uneyama, K.; Noritake, C.; Sadamune, S. J. Org.Chem. 1996, 61, 6055-6057.
27) Uneyama K.; Watanabe, H. Tetrahedron Lett. 1991,32, 1459-1462.
28) Watanabe, H.; Hashizume, Y.; Uneyama, K.Tetrahedron Lett. 1992, 33, 4333-4336.
29) Amii, H.; Kishikawa, Y.; Kageyama, K.; Uneyama, K.J. Org. Chem. 2000, 65, 3404-3408.
30) Amii, H.; Kohda, M.; Seo M.; Uneyama, K.Chem. Commun. 2003, 1752-1753.
31) Suzuki, A.; Mae, M.; Amii, H.; Uneyama, K. J. Org.Chem. 2004, 69, 5132-5134.
32) Abe, H.; Amii, H.; Uneyama, K. Org. Lett. 2001, 3, 313-315.
33) Isobe, M.; Takagi, J.; Katagiri, T.; Uneyama, K.submitted for publication.
34) Amii, H.; Kishikawa, Y.; Uneyama, K. Org. Lett. 2001,3, 1109-1112.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号