配位吸附
- 2012-12-11
- 专题
配位吸附(也称配体吸附) 是将具有配位性能的金属离子载于树脂上,通过金属离子与吸附质(配体性质的化合物) 间的配位作用而将吸附质吸附在树脂上。具有配位吸附功能的树脂称为配位吸附树脂。由于配位吸附大多在水介质中进行,而水是一种弱的配体,会与树脂上的金属离子发生较弱的配位作用。当树脂在吸附其它配体时,水从金属离子上被置换下来,因此,配位吸附又称为配体交换吸附[1,2],配位吸附树脂也被称为配体交换树脂。配位吸附的研究已经历了半个世纪的历程,现将配位吸附的研究作全面的介绍。
1 配位吸附发展简史
1952年,美国的Walton和他的学生 Stokes,将Cu2+和Ag+分别载于磺酸型和丙烯酸型离子交换树脂上,并研究了负载Cu2+和Ag+之后的树脂对水中的氨、正丁胺、 哌啶、苯胺和乙二胺的吸附情况。1953年,他们在美国化学学会第123 届会议上公布了部分结果,并于次年将完整的结果公开发表[3]。但他们的这一研究结果在以后的近 10年时间里并未引起多大的反响。1961年美国的Helfferich发现了配体交换树脂在色谱分离上的重要应用,并在《Nature 》上发表了关于配体交换色谱技术的文章[1]。接着他研究了配体吸附和配体交换的平衡,从而奠定了配位吸附的热力学基础[4]。随后大量的关于配体交换色谱的研究结果发表在《J. Chromatography 》等杂志上。被分离的物质包括氨基酸[5]、核酸和核苷酸[6~8]、生物碱[9,10]、芳胺[11~15]、脂肪酸[16]、醇和糖[17]、脂肪胺[18,19]和硫化物[20]等。上个世纪六七十年代是配体交换色谱研究最活跃的时期,70年代末期这方面的论文数量开始减少。就在配体交换色谱的研究高潮尚未过去的时候,70年代初期,另一位离子交换与吸附树脂研究领域的杰出人物,后交联技术的发明者,前苏联的Davankov开创了将配体交换色谱应用于手性氨基酸异构体拆分的应用领域。他将手性氨基酸的一个异构体,如L-脯氨酸,接到氯甲基化的聚苯乙烯树脂上,再通过L-脯氨酸与Cu2+间的配位作用而将Cu2+载于树脂上。当含有DL-脯氨酸外消旋体的溶液流过此树脂的吸附柱时,D-脯氨酸被优先吸附在柱上,而L- 脯氨酸的吸附很弱,能直接流过色谱柱,或用较低浓度的氨水即被洗脱下来。D-脯氨酸的洗脱需要用浓度较高的氨水[21,22]。采用这种手性配体交换树脂很容易将手性氨基酸对映体分开。将一系列手性氨基酸或非氨基酸手性物质载于树脂上,再负载不同的金属离子,可以实现对多种手性氨基酸的拆分[23~27]。Davankov开创了这一领域之后,大量学者开始从事这方面的研究工作。经过约10年左右的时间,到上世纪80年代后期,手性配体交换树脂对手性氨基酸拆分的研究报道逐渐减少。
上世纪90年代初到现在的10年时间里,配位吸附方面的文献报道主要集中在如下几个方面:(1)采用配位吸附来富集和回收废水和海水中微量的砷化物[28~31];(2)配位吸附和电荷作用相结合,去除和回收水溶液中微量的负离子[33~36];(3) 采用配位吸附树脂分离胺类化合物[14,15]。
2 配位吸附的优缺点
2.1 配位吸附的优点
2.1.1较高的吸附强度 由于配位键的强度较高,配位吸附也可以达到较高的吸附强度[1,2]。
2.1.2 较好的吸附选择性 配位吸附树脂只能吸附含配位原子的化合物。因此,它可以很容易地从不含配位原子的化合物中分离出具配位能力的化合物。另外对于含配位原子的化合物,由于其本身配位能力的差异,或配位时空间位阻的不同,配位吸附的选择性也出现很大的差别。因而采用配位吸附有时很容易将许多结构和性质较相近的化合物分离开来。
2.1.3 容易洗脱 一般说来,氨水可将绝大多数被吸附的配体很容易地洗脱下来,这是由于氨本身是一种强的配体,容易将吸附质置换下来。由于氨极易挥发,一般情况下被洗脱下来的吸附质经过干燥即可除去其中的氨[4]。不同的配体,由于它们在结构上的差异,被吸附的强度也有一定的差异,调节氨水的浓度可将吸附强度不同的各种吸附质依次洗脱下来。
2.2 配位吸附的缺陷
2.2.1 金属离子的脱落 在水介质中,被负载的金属离子会不同程度地从树脂上脱落下来进入吸附介质或洗脱液中,脱落的原因是水中不可避免地存在一些正负离子,其中的阳离子会与树脂上负载的金属离子发生交换,从而使被负载的金属离子从树脂上脱落下来。采用对金属离子结合能力较强的树脂,如螯合树脂作配位吸附树脂的基体,可以减少金属离子脱落的可能。金属离子的脱落会带来不利的后果:(1)会使被洗脱的物质受到金属离子的污染,需要进一步处理,以去除这些金属离子;(2)金属离子的脱落会使树脂上负载的离子不断减少,使用的过程中或使用一段时间之后,树脂上的金属离子须不断地补充,这会给实际的分离操作带来一些麻烦。
2.2.2少数配位吸附树脂的化学稳定性差 如载Fe3+的树脂在吸附苯胺时,会将苯胺氧化成苯胺黑。这种物质不溶于水,会使树脂的孔被堵塞[14]。苯胺的氧化是由于Fe3+具有一定的氧化性,Fe3+在氧化苯胺的同时变成Fe2+。
另外,有些金属离子如Ag+和Ni2+等价格较昂贵,从而限制了它们的大规模应用。
3 配位吸附树脂的基体
负载金属离子的树脂称为配位吸附树脂的基体。不同的基体对金属离子的结合能力不同。不同基体的配位吸附树脂,在负载上同样的金属离子后,对同一种配体的吸附能力也会有一定的差异。
3.1 磺酸型树脂基体
磺酸型的基体具有较高的机械强度,负载了金属离子后对配体的吸附能力很强,因为磺酸型树脂对金属离子的结合仅靠静电相互作用,被负载的金属离子上的配位位点没有被树脂基体占据,金属离子的配位位点可全部用于配位吸附。但磺酸型基体的树脂结合金属离子的强度不高,被负载的金属离子容易脱落。应用实例有:载Al3+的磺酸型树脂对DNA的分离[7];载Cu2+和Fe3+的磺酸型树脂对多元醇和糖的分离[17];载Ni2+的磺酸型树脂对乙醇胺的分离[19]等。
3.2 丙烯酸型树脂基体
丙烯酸型基体结合金属离子的强度要比磺酸型基体大得多,被负载的金属离子不容易脱落。但这种基体的树脂对配体的吸附能力比磺酸型基体的树脂弱得多,因为羧基上的氧与被负载的金属离子间有配位作用,从而占据一定的配位吸附的位点。另外丙烯酸型树脂基体的机械强度比较差,在高效液相色谱中的应用受到一定的限制。其实例有:载Cu2+的丙烯酸树脂分离硫化物[20]和安非它明药物[10];载Cu2+和Zn2+的丙烯酸树脂分离二胺和多胺[18];载Ni2+的丙烯酸型树脂分离乙醇胺;载Cu2+和Zn2+的丙烯酸树脂对氨基酸的分离[5]等。
3.3 氨基乙酸螯合型树脂基体
这种基体对金属离子的结合非常强,被负载的金属很少脱落,可在较宽的pH 范围内使用。如载Cu2+的氨基乙酸树脂对硒酸根的富集[34]和对吗啡等生物碱的色谱分离[18];载Ni2+的氨基乙酸型树脂对砷酸根和亚砷酸根的富集和回收[31]等。
3.4 键合手性氨基酸的树脂基体
这种树脂负载了金属离子后,主要用于手性氨基酸的拆分。树脂的骨架有聚苯乙烯类[21,22]、聚丙烯酰胺类[36]、聚丙烯酸酯类[37]、聚乙烯胺类和硅胶类[38]等。其中聚苯乙烯类具广谱性,能对20多种手性氨基酸进行拆分,但由于骨架亲水性不太好,拆分过程缓慢。丙烯酰胺型骨架亲水性好,明显提高了拆分效率。丙烯酸酯类骨架亲水性也较好,且结合Cu2+离子非常牢固,分离过程中几乎无铜脱落。聚乙烯胺型骨架亲水性最好,可在较短的时间完成拆分过程,且对芳香氨基酸的拆分效果尤佳。硅胶类基体的手性配体交换树脂不仅分离的速度快,效率高,而且在多种氨基酸混合物的拆分方面,尤显独特性能[26]。
4 配位吸附树脂上负载的离子
配位吸附树脂上负载的离子主要有Cu2+、Ni2+、Co2+、Fe3+、Fe2+、Ag+、Cr3+、Cd2+、Zn2+和Al3+等。选择何种离子,需考虑树脂基体对该离子的结合强度及负载量、离子对被吸附配体的结合能力以及离子本身的稳定性等多方面的因素。但目前树脂上负载的离子对配位吸附的影响没有详细的文献报道。配位吸附时选择何种离子载于树脂上主要是靠经验确定的。
Cu2+被树脂基体结合的强度高,而且结合到基体上后对配体的吸附能力也比较强[2,39]。同时它方便易得,性质比较稳定,载Cu2+的配位吸附树脂在应用研究方面的报道最多[2,3,9,10,21,22]。
载Ni2+的配位吸附树脂主要用于乙醇胺[19]、苯丙胺类药物[10]和脂肪酸[16]的分离。通常磺酸型树脂结合金属离子的强度比其它类型基体的树脂要弱,但它对Ni2+的结合强度却非常高[39]。
载Fe3+的配位吸附树脂主要集中在废水处理和污染物的控制方面。如载Fe3+的配位吸附树脂对废水或海水中砷化物的富集和回收[29~32]、载Fe3+的磺酸型树脂对芳胺和酚类物质的去除和回收[14,15]、载Fe3+的螯合型树脂对异硫氰酸盐的去除和回收[35]等。也有少量载Fe3+的配位吸附树脂在色谱分离上应用的报道,如载Fe3+磺酸型树脂对糖和多元醇的色谱分离[17]等。Fe3+的稳定性较差,遇还原性配体时易被还原成Fe2+。如以载Fe3+的配位吸附树脂吸附苯胺时,苯胺被氧化成难溶于水的苯胺黑并附着在树脂上,堵塞了树脂的微孔,因此载Fe3+的配位吸附树脂在应用上受到限制,只能吸附化学性质很稳定的配体。载Fe3+的配位吸附树脂以碱性的洗脱液洗脱时,Fe3+会变成Fe(OH)3 凝胶而牢固地被附着在树脂上,而Fe(OH)3 凝胶对配体同样具有较强吸附能力,不会影响树脂的使用性能,而且随着时间的延长,树脂的吸附能力反而增强[29~31,34]。到最后Fe(OH)3 凝胶在树脂上附着太多才使吸附量减小,这时需要用酸将Fe(OH)3 凝胶从树脂上洗尽并以FeCl3 再生树脂。由于Fe3+会使有的配体氧化,有时考虑用Fe2+载于树脂上吸附相应的配体,如载Fe2+的磺酸树脂对芳胺的吸附[15]。Fe2+虽然不会氧化被吸附的配体,但Fe2+本身容易被空气或水中的氧氧化成Fe3+,因而本身也不稳定,必须在制备树脂的当日使用,因而载 Fe2+的配位吸附树脂在应用上受到更大的限制,报道很少。
载Co2+的配位吸附树脂主要是将Co2+载在官能团化的多孔花粉素上,用以分离芳胺及核苷酸和核酸[6,11,12]。载有其它几种金属离子的配位吸附树脂仅见个别报道,如载Al3+的磺酸型树脂分离DNA[7,8]、载Cd3+的硅胶分离芳胺异构体[13]、载Zn2+的配位吸附树脂分离二胺和多胺[18]、载Cr3+的配位吸附树脂富集柠檬酸和酒石酸[32]、载Ag+的配位吸附树脂吸附二胺和哌啶[3]等。
5 配位吸附的选择性
配位吸附树脂对不同配体的吸附选择性主要取决于以下四个因素。
5.1 空间位阻[40]
对最常见的配体胺类而言,伯胺被吸附的强度最大,仲胺次之,叔胺最小。这是由配位吸附时空间位阻的不同造成的。另外,空间位阻的影响不仅取决于氮上取代基的大小和数量,还取决于与氮邻近的碳上取代基的数目。
5.2 配体的碱性
通常配体的碱性越大,被吸附的强度也越大。如考察不同碱性的取代苯胺在载Cd 硅胶柱上的容量因子,发现碱性越大的取代苯胺在载Cd硅胶柱上保留时间越长[13]。
5.3 空间构象
在配体交换树脂对手性氨基酸的拆分中,就是利用不同空间构象的氨基酸在手性配体交换树脂上被吸附的强度不同来进行的(详见7.2)。另外,在载Ca2+的配位吸附树脂分离糖类化合物时,树脂对不同糖的吸附强度与糖分子上的羟基在单糖单元环上的不同构象有关。Goulding 证实,糖的羟基以a-e-a 键形式结合在环上,更易与Ca2+配位,具这种构型的糖其吸附强度更大[40]。
5.4 配体分子中所含的配位原子数
在配位吸附树脂对多胺的分离中,配体含氮原子数越多则吸附得越牢固[18]。
5.5 介质的pH
在载Fe2+的配位吸附树脂对几种芳胺的吸附中,酸性介质有利于配位吸附。这是因为在酸性介质中,Fe2+易呈离子状态,这对配位吸附更为有利[15]。
6 配位吸附的热力学和动力学
6.1 热力学
对于一般的吸附,其热力学方程主要有Langmuir 吸附方程(C/q= C/qm+1/Kdqm,其中C为吸附质平衡浓度,q为树脂的吸附量,qm为树脂的饱和吸附量,Kd为键合常数)和Freundlich 吸附方程(q=kC1/n,其中k和n均为常数)。这两个方程对配位吸附也基本适用。尤其是Langmuir 吸附方程更能准确地描述配位吸附的平衡[11,15,29~31]。
Chanda等曾推导了针对配位吸附的平衡方程[14,15,34],即:
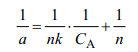
可以看出,这个方程与Langmuir吸附方程有完全相同的形式,但各项参数的意义不同,其中a为树脂上每mol金属离子吸附配体的数量,n为每mol金属离子吸附配体的饱和吸附量,CA为树脂的饱和吸附量,k为配位键合常数。以1/ a 对1/ CA作图,可直接求出树脂上每mol金属离子吸附配体的饱和吸附量及吸附的配位键合常数k[14,34]。如载Fe3+的配位吸附树脂在吸附SCN-时,树脂上的每个Fe3+可定量地与三个分子的SCN-结合[34]。
6.2 动力学
对于一般的吸附,其动力学过程与离子交换的动力学过程基本相似,吸附的动力学模型和方程基本上是在离子交换动力学的基础上作了一定的修正。离子交换的动力学过程在很早以前就被深入地研究过。一般认为离子的扩散过程是离子交换的控速步骤( 包括膜扩散和粒扩散)[41]。许多研究者,如Boyd[41]、Levenspiel[42]、Helfferich[43]和Kressman[44]等都提出过判断速率控制过程的方程或方法,其中Helfferich提出的膜扩散/ 粒扩散的简单判断方法[43](即考察不同初始浓度的F(t )- t曲线。如曲线基本重合则为粒扩散,如曲线有差异,即有初始浓度依赖性,则为膜扩散)和Kressman 提出的判断膜扩散/ 粒扩散控制的“间断法”因简单易行,被广泛应用[14,29,30,34]。其它方法因涉及的参数太多,有的参数只是估计的近似值,或实验条件与建立方程的条件有差别,只能作为判断的参考方法,用得不多。对于大多数配位吸附来说,粒扩散是主要的控制步骤,如载Fe3+的配位吸附树脂对砷化物[29,30]和酚类[14]的吸附,载Co2+的配位吸附树脂对芳胺的吸附[11]等。但也有少数膜扩散控制配位吸附的报道,如载Fe3+的配位吸附树脂对硫氰酸盐的吸附[34]等。
7 配位吸附的应用
7.1 色谱方面的应用
配位吸附的应用主要在配体交换色谱方面,这方面的应用研究报道最多,被分离的物质种类繁多,如生物碱[9,10]、其它胺类[10~15,18,19]、醇和糖[17]、脂肪酸[16]、氨基酸[5]、硫化物[20]及核酸和核苷酸[6~8]等。这些被分离的物质依靠分子中的氮、氧或硫原子与载体上的金属离子配位,并利用其空间位阻、碱性和配位原子数目等因素的差异来进行分离。配体交换色谱的出现使原来难以分离的许多化合物很容易分开,因而大大丰富了色谱分离的内容。和常规色谱分离相比,配体交换色谱具有分离选择性高、成本低、速度快的特点。配体交换色谱在分析上虽有独特的优势,但在色谱制备上存在明显的不足。因为载体上的金属离子会或多或少地脱落,分离的产物需进一步纯化,以去除混杂的金属离子。
配体交换色谱以液相色谱为主,也可以用气相色谱或薄层色谱的方式进行分离。
7.2 配位吸附树脂对手性氨基酸异构体的拆分
手性配体交换色谱仍属配体交换色谱的范畴,但由于其机理和应用独特,故在这里单独介绍。手性配体交换色谱主要用于手性氨基酸异构体的拆分。Davankov首先开创了将这一领域,30多年来,它一直是手性氨基酸异构体拆分的主要方法。手性配体交换树脂对氨基酸对映体的吸附强度的差异与树脂上担载的手性氨基酸有直接的关系。Davankov[23]提出手性配体交换树脂识别氨基酸对映体的模型,用以解释手性配体交换树脂对手性氨基酸对映体的识别机理。不管是哪种模型,有一点可以肯定,就是手性氨基酸的一对映体与树脂上的Cu2+配位稳定,而另一种对映体则因为空间上的拥挤而不太稳定。这种配位稳定性的差异决定了手性氨基酸的不同异构体在配位吸附树脂上吸附强度的不同,从而很容易通过色谱的方法将它们分离开。传统的物理法、化学法以及酶法等拆分氨基酸对映体的缺陷是效率低,需多次重复操作,且所用的手性制剂及酶制剂一般难以得到,不便回收及反复使用。采用通常的气相色谱法和液相色谱法拆分,一般需要柱前样品衍生化,费时麻烦,效率低;而手性配体交换色谱可直接拆分氨基酸对映体,无需进行柱前衍生化,分离速度快,且选择性高,流动相多采用水,实验成本低。目前手性配体固定相多采用在硅胶上键合手性分子再载以金属离子来制备,使之既具有较强的机械强度,又具有适当的亲水性,以提高色谱分离的速度。手性配体交换色谱在色谱制备上同样存在不足。除上述金属离子脱落的原因外,手性配体交换色谱的固定相对样品的处理量不大,也大大影响制备的效率。因为在载体上担载手性分子后,载体的比表面和孔容都将大大减小。如比表面为334.1m2/g,孔容为0.82mL/g 的硅胶,枝接上L-羟脯胺酸后比表面减至154.6m2/g,孔容减至0.40mL/g。比表面和孔容的减小将大大减少对硅胶样品的处理量。
7.3 废水处理及污染物的控制
水处理是配位吸附应用的重要领域之一。由于配位吸附具有很高的选择性,它可以有效地去除其它方法难以去除的水污染物[45~47]。一种通过赖氨酸臂将两个乙酸基连接在聚苯乙烯骨架上的螯合树脂[31],络合 Fe3+的强度非常高,能在极宽的 pH 范围内使用。这种树脂对废水中的污染物砷酸根及亚砷酸根具有很强富集性能。在 pH为3.5 时对砷酸根的吸附量为0.74mmol/g ,在pH为9 时对亚砷酸的吸附量为0.84mmol/g。由于对这两种不同价态砷化物吸附的最大pH 不同,可将五价砷和三价砷分开。树脂可用0.1mol/L 的NaOH再生,只有0.1%的Fe3+从树脂上泄漏,而两种砷化物可完全被洗脱。另外,载Fe3+的XFS-4195树脂、载 Fe3+的Chelex100树脂及载Fe3+的UR-10螯合树脂等,均能很好地吸附砷化物,能将海水中的微量砷富集100~200倍[28~30]。芳胺和酚类是另一类重要的污染物。这两类污染物的去除已有大量的文献报道,但配位吸附的方法是一种选择性更高的方法。载Co2+的配位吸附树脂对芳胺类化合物具有很好的吸附能力[11],载Fe2+的磺酸树脂对各种芳胺均具良好的吸附性能,而且用稀盐酸可有效地洗脱[15]。由原煤生产液化气的工业废水中含几百ppm的硫氰酸盐,它可被载Fe3+的螯合树脂XFS-4195有效地去除,树脂对SCN-的吸附量达75mg/g,而且很容易被稀碱液洗脱,洗脱时无Fe3+泄漏,树脂可反复使用。
7.4 配位吸附树脂对阴离子的富集和回收
虽然离子交换色谱可将各种离子分开,但要在工业规模上有选择地富集和回收某些离子还有难度,尤其是在其它竞争性离子浓度较高时。采用配位吸附的方法可能会有选择地吸附和分离那些可与金属离子配位的离子,如磷酸根和邻苯二甲酸根等。但单纯的配位吸附对这些离子型的配体不具有吸附分离效果,配位吸附通常只针对非离子型的物质。因此,为了有选择地吸附分离这类配体型的离子物质,需将配位吸附和离子交换两者结合起来,即将金属离子担载在螯合树脂上( 而不是离子交换树脂上),则担载的金属离子与待吸附的离子型配体间不仅存在配位作用,也存在正负电荷之间的作用,其中配位作用决定了树脂对配体型离子的选择作用,而正负电荷平衡得以使配位吸附能够实现[35]。采用这种方法可有效地在其它竞争性离子浓度较高时有选择性地吸附酒石酸、柠檬酸[32]和磷酸根[48]、砷酸根和亚砷酸根[33,35]等阴离子。采用碱-盐混合溶液或酸-盐混合溶液能有效地将被吸附的配体型离子洗脱下来。
8 配位吸附的发展趋势
配位吸附的发展经历了近半个世纪的历程,目前手性配体交换色谱仍是其应用的主要领域。
这一领域未来的发展方向是,提高手性键合固定相中金属离子的固载量,以提高色谱分离的效果,并使色谱分离向制备或半制备的方向发展;在载体上固载新型手性分子,以提高分离的选择性,或降低手性固定相制备的成本。
配位吸附的材料目前主要被制成球形颗粒用作色谱分离的固定相或其它分离材料。它也可以被制成膜的形式,使其应用领域得到进一步扩展。这方面的研究处于刚起步阶段,如将L- 脯胺酸连接到交联聚乙烯醇膜上,再载以铜离子,可有效地拆分多种手性氨基酸异构体。近年来膜分离技术已越来越成熟,并已获得了广泛的应用。随着膜技术的发展,配位吸附材料未来可能会更多地被制成膜的形式,并在更多的领域内发挥作用。
天然产物中有许多活性成分如生物碱、多酚类及多糖类,都具有配体性质,因而原则上可以采用配位吸附进行分离。如一种载Zn2+的配位吸附树脂能有效地吸附分离维生素B12。将铜离子载于磺酸树脂上,能从干苔中分离纯化干苔多糖。这种干苔多糖具有抗肿瘤活性,并能与固定化藻蓝蛋白协同作用以提高其抑制癌细胞的效果。采用配位吸附从天然产物中分离活性成分的一大优势是吸附可以在有机介质中进行。大多生物活性成分在进行吸附分离前均需经有机溶剂提取。如采用常规吸附分离,需回收有机溶剂,使活性成分的有机溶液转化成水溶液,这一转化不仅增加了分离工艺的复杂性,更重要的是造成相当部分的活性成分的损失。而配位吸附可以直接在有机介质中进行,因为配位吸附通常不会被有机介质所抑制[49]。另外,有机介质中的配位吸附还能有效地防止金属离子的脱落。随着非水介质中配位吸附研究的不断发展,配位吸附可望在天然产物分离方面得到进一步的应用。
参考文献
[1] F Helfferich. Nature, 1961, 189: 1001~1002.
[2] H F Walton. Ind. Eng. Chem. Res., 1995, 34: 2553~2556.
[3] R H Stokes, H F Walton. J. Am. Chem. Soc., 1954, 76: 3327~3330.
[4] F Helfferich. J. Am. Ch em. Soc., 1962, 84: 3237~3242.
[5] M Doury-Berthod, C Poitrenaud, B Tremillon. J. Chromatogr., 1979, 179: 37~39.
[6] E Pehlivan, S Yildiz. Sep. Sci. Technol., 1994, 29: 887~889.
[7] R M Kothri. J. Chromatogr., 1970, 52: 119~123.
[8] R M Kothri. J. Chromatogr.,1971, 56: 151~156.
[9] E Murgia, H F Walton. J. Chromatogr., 1975, 104: 417~419.
[10] C M Hernandez, H F Walton. Anal. Chem., 1972, 44(6): 890~894.
[11] M Ersoz, U S Vural, S Yildiz. Sep. Sci. Technol., 1995, 30: 3555~3557.
[12] E Pehlivan, U S Vural, A Ayar et al. Sep. Sci. Technol., 1996, 31(11): 1643~1646.
[13] D Kunzru, R W Frei. J. Chromatogr. Sci., 1974, 12: 191~196.
[14] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1983, 1: 183~189.
[15] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym. 1984, 2: 279~285.
[16 R Bedetti, V Carunchio, A Marino. J. Chromatogr., 1974, 95: 127~129.
[17] V Shaw, H FWalton. J. Chromatogr., 1972, 68: 267~269.
[18] J D Navratil, H F Walton. Anal. Chem., 1975, 47: 2443~2446.
[19] K Shimomura, T J Hsu, H F Walton. Anal. Chem., 1973, 45(3): 501~505.
[20] J W Vogh, J Edooley. Anal. Chem., 1975, 47: 816~818.
[21] V A Davankov, S V Rogozhin. J. Chromatogr., 1971, 60: 280~283.
[22] V A Davankov, S V Rogozhin, A V Semechkin et al. J. Chromatogr., 1973, 82: 359~364.
[23] V A Davankov, Y A Zolotarew. J. Chromatogr., 1978, 155: 285~288.
[24] Y A Zoloterew, N F Myasoedov, V A Penkina. J. Chromatogr., 1981, 207: 231~235.
[25] V A Davankov, S V Rogozhin, A V Semechkin et al. J. Chromatogr., 1974, 93: 363~366.
[26] D Charmot, R Audebert, C Quivoron. Liq. Chromatogr., 1985, 8: 1769~1774.
[27] D Charmot, R Audebert, C Quivoron. Liq. Chromatogr., 1985, 8: 1753~1768.
[28] I Yoshida, K Ueno, H Kobayashi. Sep. Sci. Technol., 1978, 13: 173~176.
[29] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1988, 7: 251~255.
[30] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1988, 8: 85~89.
[31] H Matsunaga, T Yokoyama, R J Edridge et al. React. Funct. Polym., 1996, 29: 167~171.
[32] Z Matajka, R Weber. React. Funct. Polym., 1990, 13: 299~302.
[33] D Zhao, A KSengupta, Y Zhu. Ind. Eng. Chem. Res., 1995, 34: 2676~2680.
[34] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1984, 2: 175~181.
[35] Y Zhu, A K Sengupta, A Ramana. React. Funct. Polym., 1992, 17: 229~234.
[36] R Audebert. J.Liq.Chromatogr., 1979, 2: 1063~1067.
[37] I A Yamskov, B B Berezin, V A Davankov. J. Chromatogr., 1981, 217: 539~543.
[38] R Audebert. J. Liq. Chromatogr., 1981, 4: 185~192.
[39] H F Walton. J. Chromatogr., 1974, 102: 57~59.
[40] R W Goulding. J. Chromatogr., 1975, 103: 229~232.
[41] G E Boyd, A W Adamson, L S Myers. J. Am. Chem. Soc., 1947, 69: 2836~2841.
[42] O Levenspiel. Chem. React. Eng.. New York: John Wiley, 1962: 343~344.
[43] F Helffrich. Ion Exchange. New York: McGrow-Hill, 1962: 324~326.
[44] T R Kressman. J A Kitcheher. Discuss. Faraday Soc., 1949, 7: 90~95.
[45] M Ersoz, M Yigitoglu, A Ayar. J. Appl. Polym. Sci., 1997, 64: 1225~1231.
[46] D Zhao, A K Sengupta. Ind. Eng. Chem. Res., 2000, 39(2): 455~462.
[47] D Zhao, A K Sengupta, L Stewart. Ind. Eng. Chem. Res., 1998, 37(11): 4383~4387.
[48] D Zhao, A K Sengupta. Water Res., 1998, 32(5): 1613~1625.
[49] J Z Li, Z Q Shi, YG Fan. Chinese J. Polym. Sci., 2002, 21: 439~444.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号