与胺/酰胺配体合作的双功能分子催化
- 2013-01-21
- 专题
1. 介绍
过渡金属类分子催化剂伴随着新型的催化转换以及人们对反应机制的了解有了进一步的发展。特别是,持续付出的大量努力开发了具有两个或者更多个活性位点协同一致的双功能分子催化剂,从而实现了有机合成高效分子变换。最近,我们开发了金属配体双功能催化剂(协奏催化剂),其中non-innocent ligands直接参与基质的激活和键的形成。双功能分子催化是一个有吸引力和总体策略的概念可以实现有效地分子改造。1)
1995年, Noyori 和他的同事们报道了二胺配体在简单的芳香酮和BINAP-Ru(II) [BINAP=2,2’-双(二苯基膦基-1,1’-联萘)]系统对映选择性加氢方面一个崭新的影响。2) RuCl2[(S)-binap](dmf)n,手性1,2-二胺和KOH组合在酮加氢反应中比经典的BINAP–Ru催化剂显示出更为高级的活性。随后,一个新的膦钌催化剂概念的雏形,以N-磺化1,2-二胺为手性试剂得到了进一步的发展,用于酮类的高效不对称转移氢化反应。3)这个真正的催化剂的详细的实验内容和理论分析表明,如Figure 1中所描述的,在循环催化过程中,具有正方形平面几何状的氨钌络合物和配位饱和的氢化(胺)钌络合物都有涉及。酰胺络合物具有足够的Brønsted碱度,很容易氢化得到含有2-丙醇基的仲醇用以生产氢化(胺)配合物。NH单元被绑定到胺配合物金属中心,具有足够的酸性可以用来激活酮基质。在氨基和胺络合物的相互转换中,金属/NH部分协同合作通过一个环状过渡态传递氢,其中,H–和H+ 被通过一个方式等量转移,从氢(胺)络合物到C=O键,或者从2-丙醇到胺络合物,并且不用金属中心的直接协调(外层机制)。双功能过渡金属分子催化剂这个独特的概念使得反应具有高利率和显著的空间立体选择性,因为 这个反应是通过一个配置紧密的设备和手性催化剂得以完成。
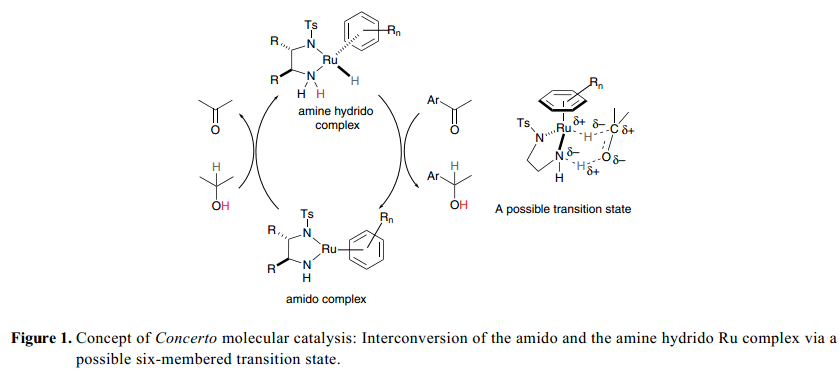
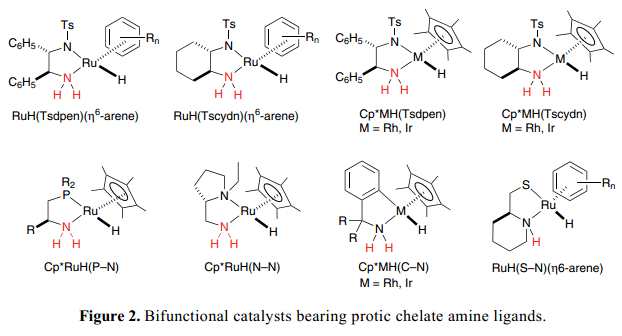
Figure 2 列举了我们组新近开发的一些有明确定义的双功能分子催化剂的代表性例子。手性双功能η6-arene–Ru络合物的概念是,RuH(Tsdpen)( η6-arene), [Tsdpen =N-(p-甲苯磺酰基)-1,2-二苯基乙二胺]已经被成功的扩展到了类似的Cp*Rh和Ir络合物中,Cp*MH(Tsdpen)[Cp*=η5-C(CH3)5,M=Rh, Ir]。4)除了N–磺酰化二胺复合物,8 和9组的金属络合物与二胺(N–N),5) 氨基膦(P–N),6) 氨基乙硫醇(S–N),7)o-氨基甲苯(C–N)8) 配体已经被广泛的开发用作完成金属/NH效应的双功能分子催化剂。
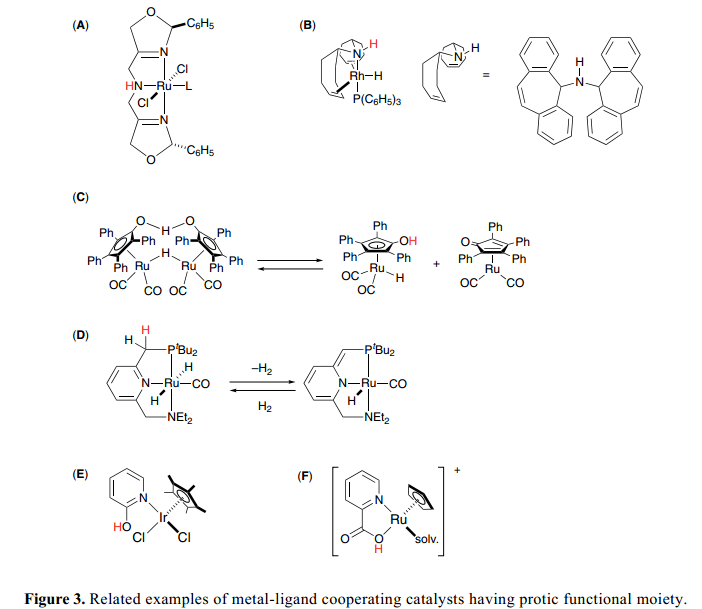
其他的研究小组研究别的相关的双功能催化剂,Figure 3表示了几组8 和9络合物,其中辅助配体上的质子官能团与金属中心协作催化氢化,氢化转移等。Zhang(A)9)和Grützmacher催化剂(B)10) 具有三叉质子胺配体,能够帮助H+/H–类似的方式下转移到双功能催化剂上面。Shvo催化剂 (C)11)可以释放催化活性物质,环戊二烯和环戊二烯酮络合物,OH基团作为质子媒介在促进氢转移过程中起到了一个关键的作用。最近,Milstein开发了新的PNN pincer complexes (D)12) 作为高活性氧化还原催化剂,并且找到了他们独特的通过脱芳构化作用的氢化/脱氢反应机制。Fujita和Yamaguchi设计了Cp*Ir 络合物(E)13),具有2-羟基吡啶和pyridonate配体,可以有效地通过酸性OH 官能团促进仲醇的脱氢反应。Kitamura和Tanaka 证明了The acidic nature of CpRu fragment (F)上的2-喹啉羧酸酸性质影响到了烯丙基醇C–O键的活性从而得到烯丙基醚。14)
本为关注于我们最新的进展,基于金属/NH协作的双功能分子催化剂,它们利用不对称还原转换反应和相关的对映选择性C–C和C–N键的生成反应。
2.使用双功能催化剂的不对称转移氢化反应1c)
一般来讲,2-丙醇可以用作一种安全、无毒、环境友好型氢源。尽管2-丙醇的不对称还原得到了一个令人满意的结果,但是,其内在的一个弊端是氢转移是可逆的,这就导致了转移的局限取决于系统的热力学因素以及反应混合物和催化剂在长时间曝光下产品对映体纯度的损耗。另一方面,甲酸遵循不对称还原反应,在不可逆转方法中转换率100%。15)
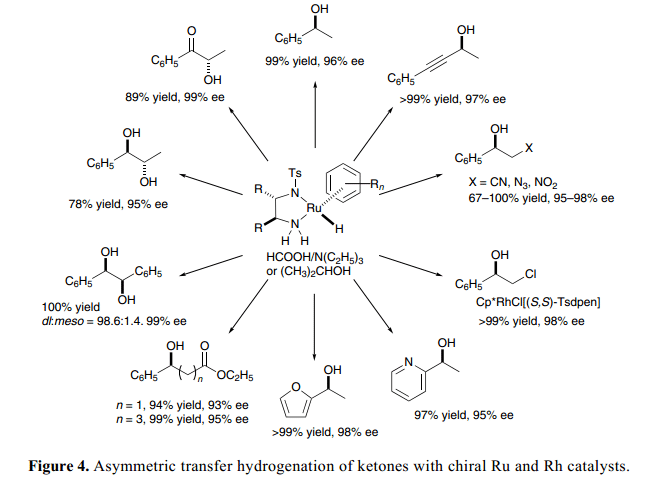
简单的芳香酮类和含有双功能催化剂的甲酸、三乙胺混合物的不对称还原反应具有高效活性、选择性、广泛的底物范围以及实用性等特点。双功能氢化(胺)-Ru配合物是配体饱和,催化系统容许有氨基、酯、氰基、硝基、叠氮化和氯基团以及芳香杂环和炔链,1c)如Figure 4总结所示。C–N双键也可以与甲酸和三乙胺含有手性Ru 或者Rh 催化剂的混合物还原得到相应的胺,具有一个很高的对映体过量百分数。16)
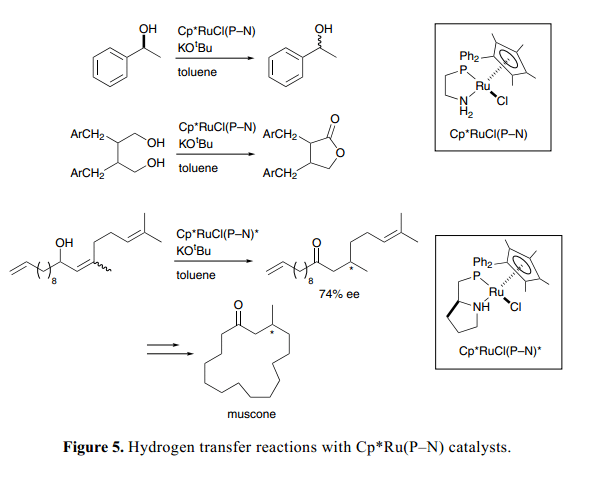
如Figure 5所示,一系列的含有质子膦胺螯合物配体的Cp*Ru(P–N)络合物 也用作氢化转移的高效催化剂。基于可逆氢化机制通过胺和氨络合物两者之间互换,氧化还原过程可以成功的用于手性醇的快速外消旋化17)以及利用丙酮将二醇特定的选择氧化为内酯。18)手性Cp*Ru(P–N)复杂络合物对映选择性异构化烯丙基醇为β-取代基酮类,接下来闭环置换得到温和的(R )-麝香酮。19)
3. 极性基质双功能催化剂的催化加氢6)
由于我们在2001年报道了醇溶剂在绑定异裂H2键到16-电子[Cp*Ru{Me2 N(CH2)2 NH2}]+(Cp*Ru(N–N))片段的的影响,20)我们系统的测试了除酮以外的极性基质的催化氢化,发现具有叔膦基替换叔胺可以导致Ru/NH 双功能范围扩大化。例如, Cp*Ru(P–N复合物,[Cp*Ru{Ph2 P(CH2)2 NH2}]+, 在各种终端环氧化物中给不饱和C–O选择性提供氢键,从而生成相应的仲醇,然而Cp*Ru(N–N) 复合物是完全消极惰性的。21)
Cp*RuCl(P–N)催化剂直接催化氢化N-acylcarbamates和carboxyimides选择性得到N -protected amines 和alcohols。22)
值得注意的是,目前的氢化作用适用于还原转化反应。双功能催化条件下伯胺从N-phthalimides脱保护同样适用于Gabriel氨基酸的合成。例如, N-邻苯二甲酰-L-苯丙氨酸甲酯通过手性N–acyloxazolidiones处理氢解得到物光学纯度损耗的L-Phe甲酯盐酸盐,这是由Evans 研发的合成手性醇和无光学纯度损耗的original 手性助剂的非常有用的合成中间体,如Figure 6所示。23a)
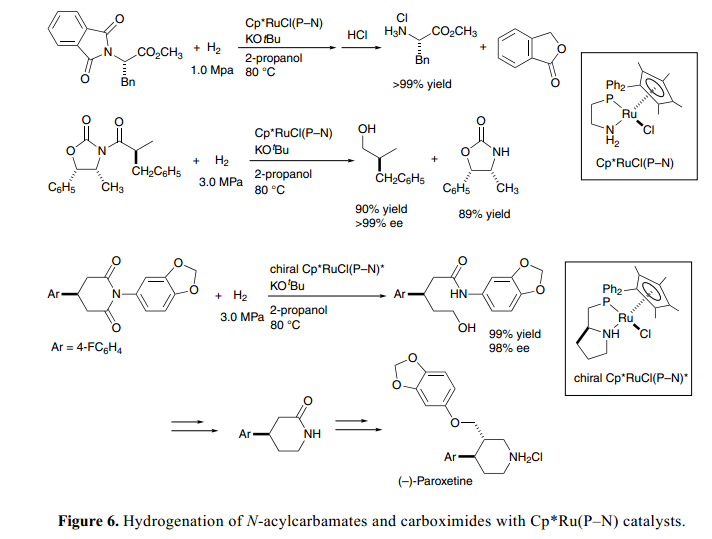
手性Cp*Ru(P–N)催化剂,具有手性P–N配合基键(由L-普氨酸衍生得到)促进前手性4-arylglutarimides对映选择性氢化通过去保护得到相对应的δ-羟基酰胺,具有很好的ee值,且产率很高。进一步细化加工得到手性哌啶酮衍生物,这个可以作用合成生理学光学活性手性化合物包括抗抑郁药物帕罗西汀等的重要合成中间体。
Cp*Ru(P–N)催化剂强压条件下也能促进酯和酰胺类氢化得到醇或者胺。23b)这个氢化过程使用金属氢化试剂化学计量法也将为传统的还原方法提供一个很强大的替代方法。
4. 双功能催化剂有氧氧化反应24)
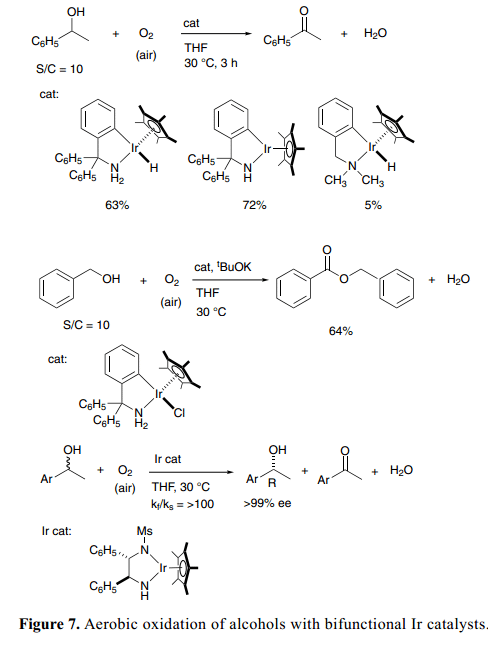
当分子氧用于双官能团醇类脱氢氧化反应中的氢原子受体时,有氧氧化变为一个简单且有机废物最小化的一个过程。幸运的是,我们发现了一个新的可孤立的官能团酰胺-Ir络合物,具有一个C–N螯合胺配体Cp*Ir[ κ2(N,C)-(NHCR2-2-C6H4)] (R=C6H5和CH3),和相应的氢(胺)络合物Cp*IrH[κ2(N,C)-{NH2CR2-2-C6H4}],可以作为酮类转移氢化反应的高效催化剂,促进催化醇类的有氧氧化。8a)1-苯基乙醇的催化反应在空气大气压强下平稳进行,含有酰胺-Ir络合物,Cp*Ir[κ2(N, C)-{NHC(C6H5)2-2-C6H4}] (Figure 7)。25)氢(胺)-Ir 络合物,Cp*IrH[κ2(N,C)-{NH2C(C6H5)2-2-C6H4}], 和二元态催化剂系统(含有氯(胺)-Rh、-Ru络合物和KOC(CH3)3), 也能得到氧化产物苯乙酮。伯醇在相同条件下反应得到二聚产品、酯类等。当含有氯络合物组分催化剂的苄醇混合物Cp*IrCl[κ2(N,C)-{NH2C(C6H5)2-2-C6H4}], 与当量的KOC(CH3)3在THF中搅拌反应,空气温度为30 °C时,得到相对应的苯甲酸苄酯衍生物。
当应用到二级醇与手性酰胺催化剂的动力学拆分时,醇的有氧氧化更为引起人们的兴趣(Figure 7)。有氧氧化条件下通过手性Ir 络合物Cp*Ir[( S,S)-Msdpen](Ms=甲磺酰),有效的解决了外消旋1-苯乙醇的动力学拆分,得到48%的 (R)-1-苯基乙醇和98% ee;kf/ ks比率为100。26)同样的,1-indanol与1-tetralol 室温下反应得到R-enantiomers,>99% ee且46–50%产率。
5.承接金属-N键的双核双功能催化剂的扩展27)
金属配体双能能效应实现了单核half-sandwich氨基络合物的形成,有望于 有效的制备具有M–N键的亚氨基-桥双核络合物。细致的结构分析和NMR研究 表明了吸电子双核集团使得亚氨基桥双核络合物稳固,28) 同时也具有一源于M–N键的酸碱双功能性质。
不饱和双(亚氨)-桥二铑(III)复合物配体,[(Cp*Rh)2(μ-NTs)2](Ts=SO2C6H4CH3-p)室温下与H2(1atm)在CH2Cl2中反应24h得到双(亚氨)-桥二铑(II)复合物(Cp*Rh)2(μ-NHTs)2],产率为82% 如Figure 8所示。应该注意的是,在这个反应中, H2形式上转换为两个氨基质子和两个电子用于二铑在[(Cp*Rh)2(μ-NTs)2]的还原。双(亚氨) 复合物与过量的2-丙醇反应得到双(胺基)复合物。两个NH质子的产生与金属中心的两个电子还原相结合与伯醇、仲醇通过不饱和单核氨基络合物脱氢作用形成了鲜明的对比,这就导致了hydrido–amine复合物的形成,并且如上所述没有形成正式的金属的还原。
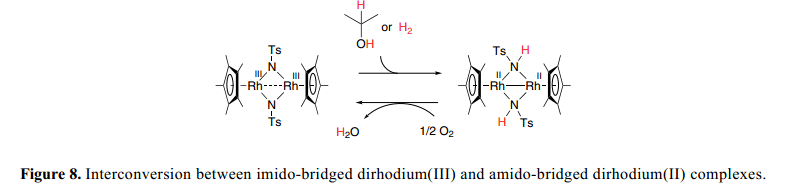
双(氨基)-桥二铑(II)复合物与O2快速反应形成双(亚氨)-桥二铑(III)复合物和水。29) 此外,双核imido和amido之间的温和的氧化还原互换可以应用到H2和醇的有氧氧化中起到催化作用。
6. 不对称共轭加成双功能催化剂30)
碱性氨基复合物可以与含酸性的有机化合物反应,得到含有金属碳键亲核试剂的胺复合物。例如,我们发现紫色Ru复合物,Ru[(R, R)-Tsdpen]( η6-三甲苯),–30°C条件下与丙二酸二甲酯在甲苯中平稳反应得到黄色结晶物,Ru[CH(CO2 CH3)2][(R,R)-Tsdpen](η6-三甲苯)。31)如果从一个氨基金属复合物中得到的具有金属键亲核试剂的胺胺复合物进攻缺电子受体,催化作用的C–C 键形成得以实现。事实上,手性Ru催化剂,Ru[(S,S)-Tsdpen](η6-arene)有效的提高了dimethyl malonate对环enones 或者nitro olefins的对映选择性Michael 加成反应得到具有很好的ee值的加成产物(Figure 9)。通过双功能Ru催化剂,1,3-dicarbonyl compounds对cyclic enones的Michael addition 反应的NMR和DFT分析结果表明对映选择性C–C键的形成通过螯合电子对中间物的形成, Ru金属中心与enone分子协同为C–C键的形成制造出一个高组织性环境,只产生一种对映选择性产品。31b)

手性氨基催化剂对映选择性共轭加成可以扩展到α –cyanoacetates与dialkyl azodicarboxylates的Ir-催化反应得到直接胺化产物。2)当乙炔酯用作亲电子试剂,α–cyanoacetate和双功能手性催化剂对映选择性和Z 选择性共轭加成得到手性加成产物,拥有一个四元碳中心,具有很好的ee值。33)
7. 总结
本文主要关注最新进展的化学概念上的新的包括立体性、区域性和化学选择性还原剂或者氧化转移以及对映选择性质C–C、C–N键(通过Michael-type共轭加成而产生)的协作催化剂与双功能过渡金属分子催化剂。配位胺试剂合理的设计使得M/NH单元中电子因素在双功能催化剂中显示平衡状态,是其具有广泛性和高实用性的催化性能的决定性因素。我们相信目前双功能分子催化剂能够为开启新的选择性分子转移包括对映选择性合成的层面上提供一个广阔的机会。
参考文献
1) a) T. Ikariya, K. Murata, R. Noyori, Org. Biomol. Chem. 2006, 4, 393. b) K. Muñiz, Angew. Chem. Int. Ed. 2005, 44 , 6622. c) T. Ikariya, A. J. Blacker, Acc. Chem. Res. 2007, 40, 1300. d) T. Ikariya, Bull. Chem. Soc. Jpn. 2011, 84 , 1.
2) a) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995, 117, 2675. b) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995, 117, 10417.
3) a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995 , 117 , 7562. b) K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya, R. Noyori, Angew. Chem. Int. Ed. Engl. 1997, 36 , 285.
4)a) K. Murata, T. Ikariya, R. Noyori, J. Org. Chem. 1999, 64, 2186. b) K. Mashima, T. Abe, K. Tani, Chem. Lett. 1998, 1199. c) K. Mashima, T. Abe, K. Tani, Chem. Lett. 1998, 1201.
5) M. Ito, M.Hirakawa, K. Murata, T. Ikariya , Organometallics 2001, 20 , 379.
6) a) M. Ito, T. Ikariya, Chem. Commun. 2007, 5134. b) M. Ito, A. Osaku, C. Kobayashi, A. Shiibashi, T. Ikariya, Organometallics 2009, 28 , 390. c) M. Ito, T. Ikariya, J. Synth. Org. Chem. Jpn. 2008, 66 , 1042.
7) a) M. Ito, Y. Shibata, A. Watanabe, T. Ikariya, Synlett 2009, 1621. b) M. Ito, A. Watanabe, Y. Shibata, T. Ikariya, Synlett 2010, 29 , 4584.
8) a ) S. Arita, T. Koike, Y. Kayaki, T. Ikariya , Organometallics 2008, 27 , 2795. b) J.-B. Sortais, V. Ritleng, A. Voelklin, A. Holuigue, H. Smail, L. Barloy, C. Sirlin, G. K. M. Verzijl, J. A. F. Boogers, A. H. M. de Vries, J. G. de Vries, M. Pfeffer, Org. Lett. 2005, 7, 1247.
9) Y. Jiang, Q. Jiang, X. Zhang, J. Am. Chem. Soc. 1998, 120, 3817.
10) a) P. Maire, T. Büttner, F. Breher, P. Le Floch, H. Grützmacher, Angew. Chem. Int. Ed. 2005, 44 , 6318. b) T. Zweifel, J.-V. Naubron, T. Büttner, T. Ott, H. Grützmacher, Angew. Chem. Int. Ed. 2008, 47 , 3245. c) T. Zweifel, J.-V. Naubron, H. Grützmacher, Angew. Chem. Int. Ed. 2009 , 48 ,
559.
11) a ) Y . B l u m , D . C z a r k i e , Y . R a h a m i n , T . S h v o , Organometallics 1985, 4, 1459. b) Y. Shvo, D. Czarkie, Y. Rahamim, D. F. Chodosh, J. Am. Chem. Soc. 1986, 108, 7400.
12) a) J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, J. Am. Chem. Soc. 2005, 127, 10840. b) C. Gunanathan, Y. Ben-David, D. Milstein, Science 2007, 317, 790. c) S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron, D. Milstein, Science 2009, 324, 74.
13) a) K. Fujita, N. Tanino, R. Yamaguchi, Org. Lett. 2007, 9, 109. b) R. Yamaguchi, C. Ikeda, Y. Takahashi, K. Fujita, J. Am. Chem. Soc. 2009, 131, 8410.
14) a) S. Tanaka, H. Saburi, Y. Ishibashi, M. Kitamura, Org. Lett. 2004, 6, 1873. b) S. Tanaka, H. Saburi, M. Kitamura, Angew. Chem. Int. Ed. 2005, 44 , 1730. c) S. Tanaka, T. Seki, M. Kitamura, Angew. Chem. Int. Ed. 2009, 48 , 8948.
15) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996, 118, 2521.
16) a) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996 , 118 , 4916. b) J. Mao, D. C. Baker, Org. Lett. 1999, 1, 841.
17) M. Ito, A. Osaku, S. Kitahara, M. Hirakawa, T. Ikariya, Tetrahedron Lett. 2003, 44 , 7521.
18) a) M. Ito, A. Osaku, A. Shiibashi, T. Ikariya, Org. Lett. 2007, 9, 1821. b) M. Ito, A. Shiibashi, T. Ikariya, Chem. Commun. 2011, 2134.
19) M. Ito, S. Kitahara, T. Ikariya, J. Am. Chem. Soc. 2005, 127, 6172.
20) M. Ito, M. Hirakawa, K. Murata, T. Ikariya , Organometallics 2001, 20 , 379.
21) M. Ito, M. Hirakawa, A. Osaku, T. Ikariya, Organometallics 2003, 22 , 4190.
22) a) M. Ito, A. Sakaguchi, C. Kobayashi, T. Ikariya, J. Am. Chem. Soc. 2007, 129, 290. b) M. Ito, C. Kobayashi, A. Himizu, T. Ikariya, J. Am. Chem. Soc. 2010, 132, 11414.
23) a) M. Ito, L. W. Koo, A. Himizu, C. Kobayashi, A. Sakaguchi, T. Ikariya, Angew. Chem. Int. Ed. 2009, 48 , 1324. b) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu, T. Ikariya, J. Am. Chem. Soc. 2011, 133, 4240.
24) T. Ikariya, S. Kuwata, Y. Kayaki, Pure Appl. Chem. 2010, 82, 1471.
25) S. Arita, T. Koike, Y. Kayaki, T. Ikariya, Chem. Asian J. 2008, 3, 1479.
26) S. Arita, T. Koike, Y. Kayaki, T. Ikariya, Angew. Chem. Int. Ed. 2008, 47 , 2447.
27) S. Kuwata, T. Ikariya, Dalton Trans. 2010, 39 , 2984.
28) a) K. Ishiwata, S. Kuwata, T. Ikariya, Organometallics 2006, 25 , 5847. b) H. Arita, K. Ishiwata, S. Kuwata, T. Ikariya, Organometallics 2008, 27 , 493.
29) K. Ishiwata, S. Kuwata, T. Ikariya, J. Am. Chem. Soc. 2009 , 113, 5001.
30) a) T. Ikariya, I. D. Gridnev, Chem. Rec. 2009, 9, 106. b) T. Ikariya, I. D. Gridnev, Top. Catal. 2010, 53 , 894.
31) a) M. Watanabe, K. Murata, T. Ikariya, J. Am. Chem. Soc. 2003, 125, 7508. b) I. D. Gridnev, M. Watanabe, H. Wang, T. Ikariya, J. Am. Chem. Soc. 2010, 132, 16637.
32) Y. Hasegawa, M. Watanabe, I. D. Gridnev, T. Ikariya, J. Am. Chem. Soc. 2008, 130, 2158.
33) Y. Hasegawa, I. D. Gridnev, T. Ikariya, Angew. Chem. Int. Ed. 2010, 49 , 8157.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号