从不对称催化剂到不对称自催化剂
- 2012-03-14
- 专题
1、介绍:
众所周知,多数生理活性物质都是由不对称碳原子组成的手性分子。随着不对称合成工艺的发展,如何进行预期的绝对构型对映体的选择性合成是有机合成专家面对的一个重要课题。特别是,以不对称催化剂的催化作用为基础的不对称合成更是一个活跃的研究领域。包括作者在内的多数研究小组的研究表明,在二烷基锌与醛的不对称烷 基化的过程中,利用手性氨基醇作为不对称催化剂,已成为光学活性仲醇领域中的主流反应 1) 。利用脯氨酸衍生的手性氨基醇、N,N-双烷基去甲麻黄碱的衍生物或手性哌嗪作为不对称催化剂,作者开发了一种具有高对映体选择性的醛的不对称烷基化工艺。在这个反应中,还可以得到高收率的通用底物和许多有用的中间体。
通过精确的分子结构设计形成一个新功能的物质也是十分有趣的。此前,还没有已知的手性分子可做为具有自我复制能力的不对称自催化剂。我们发现手性嘧啶和喹啉醇是具有高对映体选择性的不对称自催化剂,不但有自我增值的能力,而且有很好的光学纯度2)。
本文我们将介绍我们在醛类的不对称催化剂和不对称自催化剂方面的研究结果。
2、利用手性氨基醇进行醛的不对称烷基化
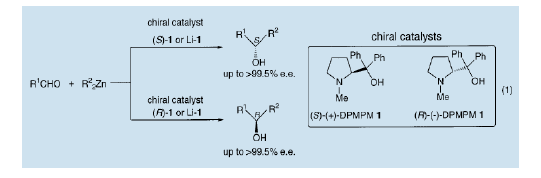
1)、利用二苯基(1-甲基-吡咯-2-基)甲醇(1,DPMPM)进行醛的不对称烷基化。
我们已经研究了利用脯氨酸及其衍生物制备的手性氨基醇(DPMPM)对二烷基锌进行的不对称烷基化3)4) 。结果在芳香醛的不对称加成反应中,相应的光学活性仲醇具有极高的光学纯度。利用DPMPM的锂醇提供的高对映体选择性进行的不对称乙基化,得到了几乎完全光学纯度的1-苯基-1-丙醇。通过对不对称催化剂绝对构型的选择,产品的S-和R-构型能够被分别制备出来。也就是说,利用(S)-(+)-DPMPM和(R)-(-)-DPMPM可以分别得到S-和R-构型的醇类。
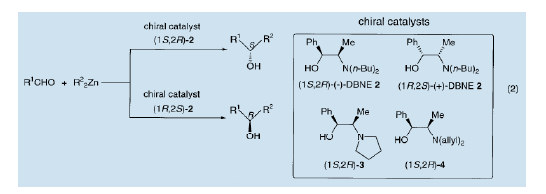
2)、利用N,N--双烷基去甲麻黄碱进行醛的不对称烷基化。
手性氨基醇,N,N-双烷基去甲麻黄碱,对醛的不对称烷基化也是一个有用的不对称催化剂5) 。一般来说,与芳香醛相比,脂肪醛的不对称烷基化很难得到高收率。我们发现N,N-双烷基去甲麻黄碱无论对脂肪醛还是芳香醛都是一个对映体选择性很高的催化剂。根据烷基取代基上的氮原子的结构不同,N,N-双烷基去甲麻黄碱也表现出不同对映体选择性。例如,在双烷基锌进行的脂肪醛的不对称乙基化中,N,N-双烷基去甲麻黄碱(2,DBNE)表现出很高的对映体选择性(e.e值为93%)5a)。在二异丙基或二丁酯锌进行的不对称烷基化中使用N,N-双烷基去甲麻黄碱也能表现出高选择性6)。此外,1-苯基-2-吡咯烷基-1-丙醇(3)和N,N-二烯丙基去甲麻黄碱(4)也是有用的不对称催化剂5b)。
3)、二乙基锌的对映选择加成到一个带官能团的醛。
利用手性氨基醇1和2可以实现各种醛的不对称烷基化(方案1)。例如,以炔醛为基板产生一个手性丙炔醇(5)7a)。当使用氘代和含氟醛时,可以分别得到大量的具有光学活性的重氢取代(6)7b) 和含氟醇(7)7c)。通过双烷基锌对醛的化学和对映选择性加成,酮醛可产生手性醇酮(8)7d)。对羰基烯酮的不对称加成,产生大量的不对称烯丙醇(9)4b,7e)。此外,手性羟醛(10)7f) 和杂环醇(11)7g)也可以被不对称地制备出来。具有高对映选择性和立体选择性的双醛加成工艺,产生高光学纯度的二醇(12)7h) 。同时带有酯基和醛基的化合物的烷基化会导致一个羟基酯的产生。该羟基酯可转化为类似于手性内酯那样的手性酯类(13)7i)。手性酞,芹菜中的一个成份,可以通过醛的不对称烷基化,从卤化苯甲醛得到,卤化基转换为酯基,并且由此产生羟基酯之后的内酯7j)。
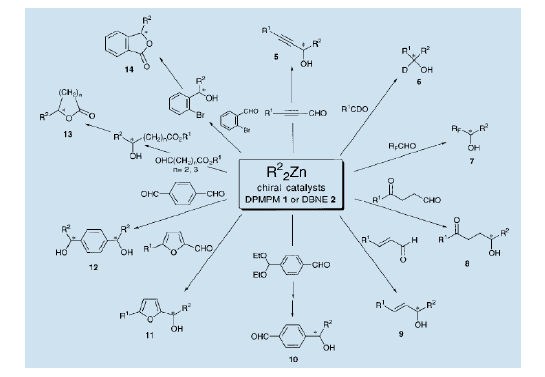
氨基醇(1R,2S)-3做为不对称配体已成为通过酮的不对称烷基化合成HIV逆转录酶抑制剂的关健一步8)。
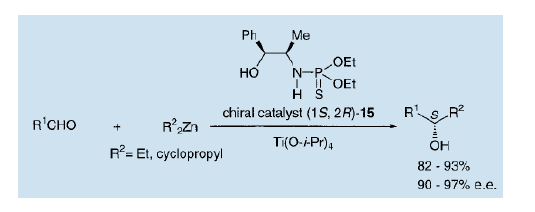
4)、利用硫代磷酰胺进行的不对称烷基化
在不对称反应中利用手性硫代磷酰胺的例子是极少的。研究表明,醛的不对称烷基化可以在四异丙基钛存在时,以硫代磷酰胺(15,PHONE)作为催化剂来实现9)。这一反应是在低温下(-45℃)进行的,可以得到高产率的不对称光学活性的仲醇。这个催化剂也可以用于通过双环丙基锌进行的醛的不对称环丙烷化,来生产光学纯度接近97%(e.e.)的光学活性的环丙醇10)。
3、不对称自催化剂
通过对吡啶-3-甲醛反应的观察,以确定用氨基醇1和2为催化剂的不对称烷基化一般性的规律。作者发现,反应速度要高于其他醛(反应时间:约3个小时,苯甲醛反应时间:约20小时)11) 。这表明,通过反应产生的化合物具有不对称自催化剂的功能。
在传统的不对称催化中,不对称催化剂C和产品D的结构一般是完全不同的。但在不对称自催化中,不对称催化剂C的结构和绝对构型与产品C完全相同。
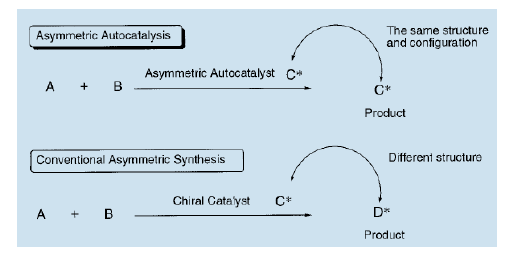
不对称自催化拥有以下三大优势:
i)、反应效率高。在不对称自催化中,反应体系中的不对称催化剂收益随着反应的进行而增加。因此,反应效率非常高,反应能迅速完成。
ii)、反应过程简单。传统工艺中要求进行的不对称催化剂与反应产物的分离,在这里是不必要的,因为它们有着相同的结构。
iii )、连续反应具备了可能性。由于产物可以充当不对称催化剂,因此,在不对称自催化体系中的催化剂可以反复地得到和补充。微量的催化剂就可以使大量的手性化合物反应得以进行。
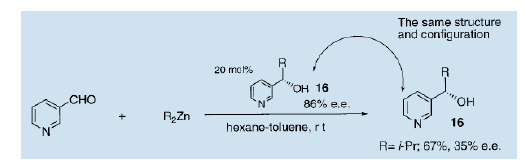
如上所述,与传统工艺相比较的优势而言,不对称自催化很可能成为下一代的节能的不对称反应。目前还没有关于不对称自催化相关的书面实验报告,虽然Wynberg已经描述了反应的意义12)。1990年,我们首次报导了在3-吡啶醛的烷基化过程中使用一定量的3-吡啶醇作为自催化剂,从而得到结构和绝对构型均相同的3-吡啶醇的自催化反应13)。利用86%(e.e.)的(S)-2-甲基-1-(3-吡啶基)-1-丙醇(16)作为不对称自催化剂,通过双异丙醇锌对3-吡啶甲醛进行的异丙烷化得到了结构和绝对构型与自催化剂完全相同的(S)-3-吡啶醇,收率为67% e.e值为35%。
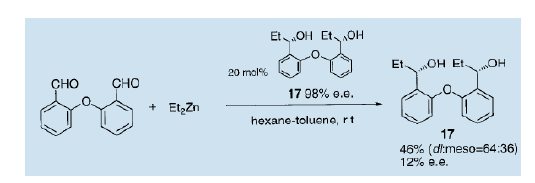
然后,我们合成了一种手性二醇并使用它作为不对称自催化剂14)。我们发现,这个带有二苯醚架构的手性二醇(17)在通过双乙基锌对相应的二醛进行的不对称二乙烷化过程中充当不对称自催化剂, 可以生产e.e值为17的二醇(17)。
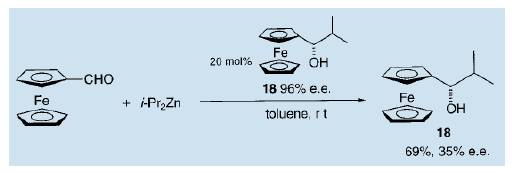
此外,我们还确定了在二茂铁基醛与二异丙基锌的反应中,使用手性二茂铁基醇(18,e.e值为96%)作不对称自催化剂,也可以得到收率为69%,不对称量为35%的相同的醇(18)15)
1)、高对映体选择性不对称催化剂
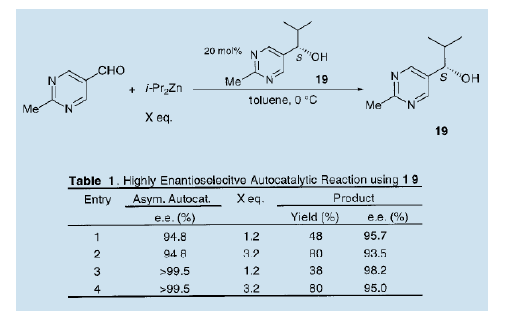
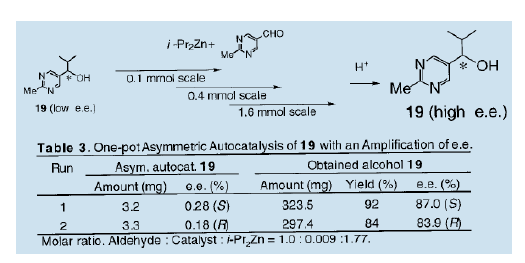
综上所述,自从我们发表第一个关于不对称自催化的报告后,不对称自催化工艺过程的发展得到了关注13)。但是,事实上仍然存在着一个悬而未决的问题,即产品的光学纯度还远低于催化剂的光学纯度。我们进一步研究,在考虑到分子对称性和醛的反应的同时,制备高对映体选择性的不对称自催化剂。结果表明,含有两个氮原子的5-嘧啶醇是一种优良的不对称自催化剂。我们详细地考察了反应并发现手性5-嘧啶醇是一种非常有效的不对称自催化剂。二异丙基锌与5-嘧啶醇的不对称异丙基化过程中,可以得到高不对称量的与自催化剂绝对构型相同的手性5-嘧啶醇(e.e值90%以上) 16)。当(S)-5-嘧啶醇(19, e.e值94%)作为不对称自催化剂时,新的产品(S)-5-嘧啶醇(19)的光学纯度是95.7% ( e.e值),这意谓着该化合物具有自我增值而不会失去其光学活性的功能。当接近完全光学纯的嚓咤醇(19)使用后,新产生的化合物(19)光学纯度高达98.2% ( e.e值)。
只含有一个氮原子的3-哇琳醇也被证明是非常高效的不对称自催化剂7)。具体地说,在3-喹啉甲醛的不对称异丙基化中使用3-喹啉醇(20, e.e值94%)作不对称自催化剂,可得到高光学纯度(e.e值为94%)的哇琳醇(20)

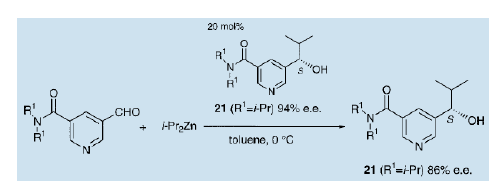
我们还发现,在5位带有一个甲酞基的3-毗咤醇作为不对称自催化剂较5位不带任何官能团的催化效果要好得多18)。不对称量的大小取决于甲酞基中氮原子上的烷基的性质。当R1是一个异丙基的3-吡啶醇(21)用于催化时,产品的不对称量约为86% ( e.e值)。
2)、不对称自催化与光学纯度的改善
Kagan等首先报导了不对称氧化的非线性光学现象,一种产品的光学纯度高于使用不对称催化的现象19a)。Ogunil9b), Noyori等19c)还就醛的不对称烷基化进行了进一步研究19d)。当然,所有的反应物都不是自催化剂,产品的结构也与不对称催化剂的不同。虽然可以从e. e值为10%的催化剂中可以得到e.e值为50%的产品,但进一步改善产品的光学纯度是不可能的。
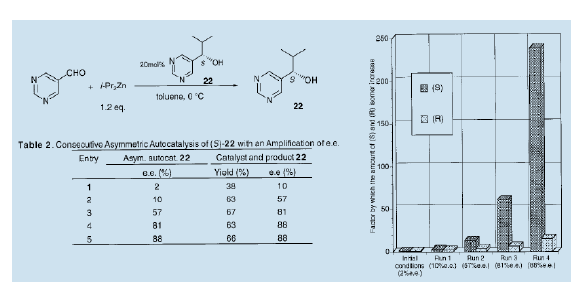
另一方面,我们发现pw陡醇是有效的自催化剂。偿试连续的不对称自催化反应,以实现大部分情况下的反应是自催化反应。首先,不对称自催化反应是利用e.e值仅有2%的(S)嘧啶醇(22)进行的。结果表明,得到的新的嘧啶醇(22)的光学纯度(产品和使用的自催化剂的总和)提高至10%(e.e值)。在下一阶段的反应中使用10% Ce.e值)的化合物作为不对称自催化剂进一步提升光学纯度,嘧啶醇(22)的e.e值可达到57%。在随后的不对称自催化反应中陆续使用获得的醇,光学纯度可进一步提升至81%(e.e值),甚至达到88% (e.e值)20)。起始时略有过量的S构型的醇陆续经过四次反应后,自身扩增了238倍,而R-构型的醇只扩增了16倍。光学纯度从2% (e.e值)扩增到88%(e.e值)。
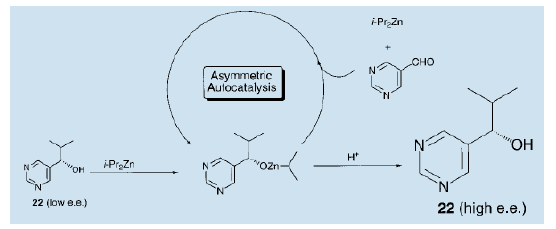
我们假设如下:嘧啶醇(22)锌是真正的不对称自催化剂。二异丙基锌与醛的加成物使不对称自催化反应得以进行,从而改善了光学纯度。反应完成后,反应物酸化调整为高光学活性的嚓咤醇(22)。
以上反应中,为了孤立嘧啶醇并用于随后的下一阶段反应中,每个反应之后,反应物都要经过酸处理。在每个循环完成之后,偿试陆续在一个容器中加入二异丙基锌和醛,以确保反应不停下来。结果表明,大大提高嘧啶醇的光学纯度是切实可行的21)。例如,在二异丙基锌和醛的反应中,使用低光学纯度0.2% ( e.e值)的催化剂,生产的产品接近90% ( e.e值)。
3)、自我复制型不对称还原
综上所述,我们发现不对称自催化发生在少数双烷基锌与醛的不对称烷基化中。那么,这种不对称来源和产物的构型和结构都相同的“自复制型反应”能否应用于其他反应?
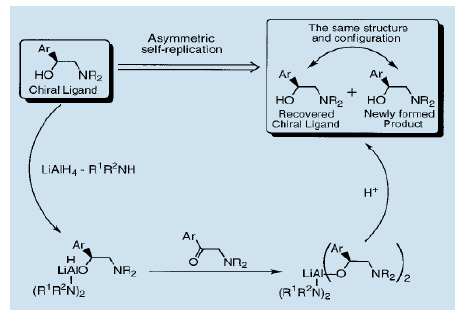
我们发现利用手性氨基醇做为配体对氨基酮进行加氢还原可以得到与那些配体结构和绝对构型完全相同的氨基醇。我们还发现利用氢化钾铝对α-氨基酮进行不对称还原是可能的。非对称β-氨基醇催化剂和非手性胺配体被用来产生一个与不对称配体结构和绝对构型完全相同的氨基醇22)。这种反应可能被视为一个自我复制型不对称还原。特别是在氮原子上带有一个杂环取代基的氨基醇,甚至更有效,可以将光学纯度提升至90% ( e.e值)。
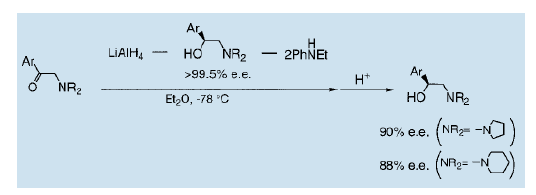
这些被重结晶的氨基醇的光学纯度得以提高。围绕重结晶的自我复制型不对称反应物建立一个手性氨基醇自我复制的完整循环。许多手性氨基醇,如上述的化合物1和2都可以作为高对映体选择性催化剂。其他有名的例子是具有药理和生理活性的化合物。这使得手性氨基醇的自我复制型不对称反应成为一个有价值的合成工艺。
4、结论
手性氨基醇,如DPMPM (1)和DBNE(2)在醛的不对称烷基化过程中,充当不对称催化剂井得到不对称量很高的具有光学活性的二醇。手性嘧啶、喹啉和吡啶醇作为不对称的自催化剂,并且是自我增值的不对称催化。此外,嚓陡醇是微量的敏感的手性不对称分子23)。这导致光学纯度的放大。
估计将来还会有其他的有趣的功能被发现。
参考文献
1.a)K.Soai,S.Niwa,Chem.Rev.,92,833(1992)
b)R.Noyori,M.Kitamura,Angew.Chem.Int.Ed.Engl.,30,49(1991)
c)K.Soai,T.Hayase,Yuki Gosei Kagaku Kyokaishi (J.Synth.Org.Chem.Jpn.),53,138(1995)
d)Aminoalcohols promote the ethylation of benzaldehyde by diethyl zinc.
T.Mukaiyama,K.Soai,T.Sato,H.Shimizu,K.Suzuki,J.Am.Chem.Soc.,101,1455(1979)
2.K.Soai,T.Shibata,Yuki Gosei Kagaku Kyokaishi(J.Synth.Org.Chem.Jpn.),55,994(1997)
3.a)K.Soai,A.Ookawa,J.Chem.Soc.,Chem.Commun.,1986,412
b)A.Ookawa,K.Soai,J.Chem.Soc.,Perkin Trans.1,1987,1465
4.a)K.Soai,A.Ookawa,K.Ogawa,T.Kaba,J.Chem.Soc.,Chem.Commun.,1987,467
b)K.Soai,A.Ookawa,T.Kaba,K.Ogawa,J.Am.Chem.Soc.,109,7111(1987)
5.a)K.Soai,S.Yokoyama,K.Ebihara,T.Hayasaka,J.Chem.Soc.,Chem.Commun.,1987,1690
b)K.Soai,S.Yokoyama,T.Hayasaka,J.Org.Chem.,56,4264(1991)
6.K.Soai,T.Hayase,K.Takai,T.Sugiyama,J.Org.Chem.,59,7908(1994)
7.a)S.Niwa,K.Soai,J.Chem.Soc.,Perkin Trans.1,1990,937
K.Soai,S.Niwa,Chem.Lett.,1989,481
b)K.Soai,Y.Hirose,S.Sakata,Bull.Chem.Soc.Jpn.,65,1734(1992)
c)K.Soai,Y.Hirose,S.Niwa,J.Fluorine Chem.,59,5(1992)
T.Hayase,T.Sugiyama,M.Suzuki,T.Shibata,K.Soai,J.Fluorine Chem.,84,1(1997)
d)K.Soai,M.Watanabe,M.Koyano,Bull.Chem.Soc.Jpn.,62,2124(1989)
K.Soai,M.Watanabe,M.Koyano,J.Chem.Soc.,Chem.Commun.,1989,534
M.Watanabe,K.Soai,J.Chem.Soc.,Perkin Trans.1,1994,3125
e)K.Soai,Y.Kawase,A.Oshio,J.Chem.Soc.,Perkin Trans.1,1991,1613
f)K.Soai,H.Hori,M.Kawahara,Tetrahedron:Asymmetry,1,769(1990)
g)K.Soai,Y.Kawase,S.Niwa,Heterocycles,29,2219(1989)
h)K.Soai,H.Hori,M.Kawahara,J.Chem.Soc.,Chem.Commun.,1992,106
i)K.Soai,S.Yokoyama,T.Hayasaka,Chem.Lett.,1988,843
j)K.Soai,H.Hori,M.Kawahara,Tetrahedron:Asymmetry,2,253(1991)
8.A.S.Thompson,E.G.Corley,M.F.Huntington,E.J.J.Grabowski,Tetrahedron Lett.,36,8937(1995)
A.S.Thompson,E.G.Corley,M.F.Huntington,E.J.J.Grabowski,J.F.Remenar,D.B.Collum,J.Am.Chem.Soc.,120,2028(1998)
9.K.Soai,Y.Hirose,Y.Ohno,Tetrahedron:Asymmetry,4,1473(1993)
K.Soai,Y.Ohno,Y.Inoue,T.Tsuruoka,Y.Hirose,Rec.Trav.Chim.Pays-Bas,114,145(1995)
10.T.Shibata.H.Tabira.K.Soai.J.Chem.Soc.,Perkin Trans.1,1998,177
11.K.Soai,S.Hori,S.Niwa,Heterocycles,29,2965(1989)
12.H.Wynberg,J.Macromol.Sci.Chem.,Sect.A,26,1033(1989)
13.K.Soai.S.Niwa.H.Hori. J.Chem.Soc.,Chem.Commun.,1990.982
14.K.Soai T.Hayase,C.Shimada,K.Isobe,Tetrahedron:Asymmetry,5,789(1994)
15.K.Soai T.Hayase,K.Takai,Tetrahedron:Asymmetry,6,637(1995)
16.T.Shibata H.Morioka,T.Hayase,K.Choji,K.Soai,J.Am.Chem.Soc.,118,471(1996)
17.T.Shibata K.Choji,H.Morioka,T.Hayase,K.Soai.Chem.Commun.,1996.751
18.T.Shibata H.Morioka,S.Tanji,T.Hayase,Y.Kodaka,K.Soai,Tetrahedron Lett.,37,8783(1996)
19.a)C.Puchot,O.Samuel,E.Dunach,S.Zhao,C.Agami,H.B Kagan,J.Am.Chem.Soc.,108,2353 (1986)
b)N.Oguni,Y.Matsuda,T.Kaneko,J.Am.Chem.Soc.,110,7877(1988)
c)M.Kitamura,S.Okada,S.Suga,R.Noyori,J.Am.Chem.Soc.,111,4028(1989)
d)C.Girard,H.B.Kagan,Angew.Chem.Int.Ed.,37,2922(1998)
20.K.Soai,T.Shibata,H.Morioka,K.Choji,Nature (London),378,767(1995)
注:本文为提供者翻译的,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号