利用横向分子间相互作用控制液晶分子超晶格结构
- 2012-04-10
- 专题
1. 介绍
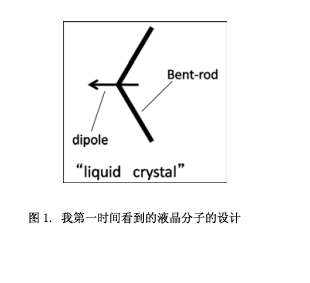
1997年的5月,那个时候我正在想“我应该怎样来改变我的化学?”从学生时代我都一直在做有机化学的研究项目,而那个时候,我是一名35岁的助理教授.我无法停止思考这个问题,怎样去扩展我的化学领域,我认为我必须得开始着手一个与我现在正在的研究方向完全不同的领域。然后我决定去美国或者是欧洲学习超分子化学或者是材料化学。然而, 那个时候我在美国或者是欧洲没有有联系的教授,因此我给在这些领域里面做得很好的专家发邮件,争取一个博士后的学习机会。第一个为我提供机会的是麻省理工学院的Timothy M. Swager教授,他是超分子化学和材料科学领域的权威,幸运的是,他回复我“你将什么时间过来?”然后我开始了麻省理工学院的学习。在第一天,Swager教授给了我一张纸,上面写了一个简单的字符 (图1),在横向方向的弯曲杆的中心有一个箭头。他说“如果你合成这类分子,就会发现有趣的现象”。这似乎是一个“液晶”的研究.随后,我查阅了英日字典意识到这个“液晶”是一个存于晶体和液体的中间状态。然而,当时我没有认识到这个新项目的价值。“什么是液晶?”知道了这个单词的含义之后这个简单的问题一直留在我的脑海里面。这是一个从零开始的研究(没有任何相关的知识)。
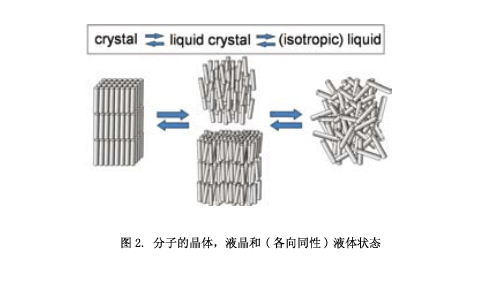
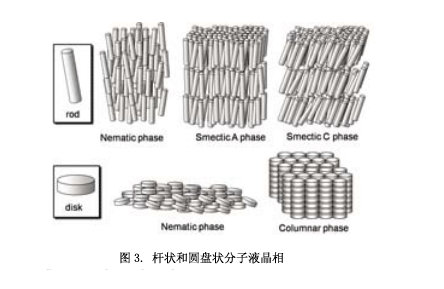
如图2所示,液晶是晶体和液体(各向同性)之间的中间状态。它兼有流动性和位置有序性的特点。杆状和圆盘状的分子具有液体的晶体。例如(图3),类似棒状分子的液晶相中,已知的有向列相、近晶A相、近晶C相;圆盘状分子中液晶相有向列相和柱状相。此外,还有三维光学各向同性的立方相。
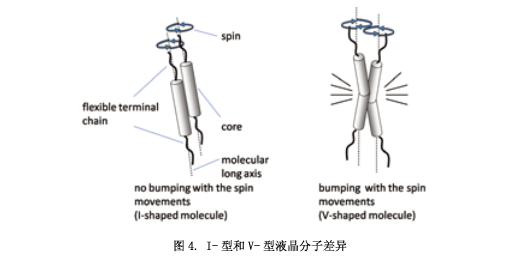
多数用于液晶显示器的液晶分子具有一个像杆状“I”的性状结构(图4左)。I状分子围绕着液晶相长轴快速旋转,这个旋转运动对液晶相的稳定有很重要的作用,通过高速的运转阻止分子在低温液晶范围内结晶。然而,曲棒状分子(V-型分子) (图4右)不能够顺利的围绕分子轴转动,从而导致液晶相的不稳定性。
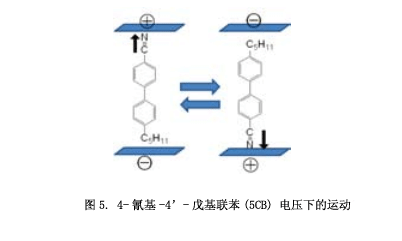
I型液晶分子在分子的末端具有一个极性取代基,这些分子会对电场作出回应。例如,4-氰基-4’-戊基联苯(5CB) 用于液晶显示器,施加电压将会改变其场方向。如图5所示 (箭头表示分子偶极),通过电场,分子平行排列。分子长轴方向上的电极性不但能够抑制分子的转动,而且会形成一个定向的分子棒状结构。然而,I型液晶分子具有一个横向的分子极性,通过分子间的偶极-偶极相互作用抑制其旋转,导致液晶相的不稳定性。特别是他的中心具有一个横向的偶极分子,不能顺利旋转,很容易重结晶。
现在,我清楚了由Swager教授设计的这个分子对于液晶相的稳定性有两处缺点(V型和中央横向偶极子) 。这种分子构型对于制备稳定的液晶相是不合理的。这是一个非常具有挑战性的项目,在麻省理工的前三个月,我合成了30种这种具有弯曲棒状和中央偶极子的分子。这些分子理所当然的没有显示出任何的液晶相态,他们很容易就会结晶了。
与此同时,大量的实验和失误之后,我发现了一个暗示,能够使具有V型和中央横向偶极子的分子生成一个稳定的液晶相。锚和螺旋转结构是产生稳定液晶相的必要条件1) 。曲杆和中心偶极像锚一样停止分子转动。但是如果分子中有螺旋结构转动,那么它保持分子运转抑制分子结晶,从而稳定液晶相1) 。螺旋结构与锚结构分离在稳定液晶相方面有很高的影响。
这个概念可以扩展延伸到其他具有强分子间相互作用的液晶分子的合成中。如果分子中的膜部分保持运转、震动、滑动,那么这个分子就不会结晶从而保持液晶状态。虽然这个概念不是很新颖的,我依旧在MIT 学习了一年的时间。在那之后, 我的项目就是选择合成具有一个大的横向影响作用的新型液晶分子的合成。这个概念基于“分子部分的持续移动”。
2. 分子中心横向方向上偶极间的相互作用的介绍
化合物1和2在MIT合成的(图6)2) 。这些分子中心部分是一个具有两个氰基基团的噻唑环,并且有一个大的偶极矩(超过6 debye的计算值)。化合物1长度较短,液晶相不稳定,其中一个向列相在26℃,低于其熔点温度时被观测到会处于一种超级冷却状态。分子的相态从液晶态瞬间转换为结晶态,因为他们具有弯曲的杆结构,短分子轴方向有一个很大的分子偶极子,分子旋转受到强烈的阻碍。然而,令我惊奇的是化合物2,在1的两个核心终端各引入一个苯环,表现出非常稳定的液晶相。它的向列相和近晶相A的相温度范围分别是160-252和135-160℃。这个液晶相的稳定性的改进主要是源于分子的终端引入了旋转结构。纵横比(=分子长度/分子的宽度)的增加,引入两个苯环和两个酯基增强了其分子间的相互作用,都被认为是其稳定性的原因。


化合物3(图7a) 是我在完成MIT 项目后,在千叶大学合成的第一个液晶化合物3) 。它具有高度弯曲的形状和一个大的偶极矩。这两个棒状介晶中心由-CO-NR-CO-(R=alkyl chain) 链接形成一个U型分子,具有12 debye的分子偶极。这个化合物在98-199℃显示出稳定的近晶相A(图7b).化合物3的X射线衍射图中, 可观测到两个图层的距离(图7c)。一般情况下,近晶相A只有一层间隔。循环加热和冷却过后,这两个隔层融合成为一个层距。这种分子具有很强的分子偶极子,分子间的相互偶极作用很强。这个层面临近的分子不能够在分子的长轴方向上顺利的转动,因为分子间强烈的相互作用。此外,如图7d所示,每个分子有两个介晶芯终端链,而化合物3在链接方向只有一个终端链。分子一端与另一端的烷基链空间平衡是不一样的。因此可以认为是这种不平衡导致产生了两种不同类型的层结构。一个相邻层之间没有有交错的结构,一个相邻层之间有交错结构。两层距离之间的差异与交叉烷基链的长度相对应。
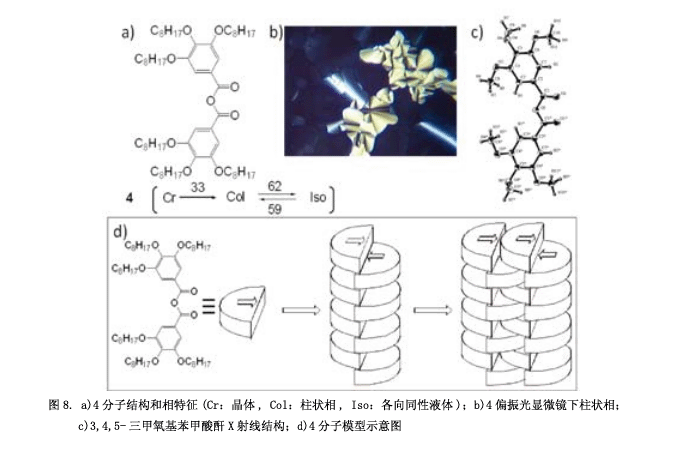
接着,一个更为简单的分子(4)被设计合成,它具有大的偶极和弯曲的棒状结构(图8a)。它显示一个柱状的液晶相(图8b) 4) 。在化合物4中的三个烷氧基能够为苯环提供电子,羰基可以阻止水分子进攻羰基碳原子。因此,化合物4是一个稳定的酸酐,可以用柱层析法纯化和重结晶。基于3,4,5-三甲基苯甲酸酐(图8c)的X射线和其分子模型,分子间强大的偶极作用使得分子聚集成为一个柱状分子团,成不平衡的并列状构型(图8d)。
3.横向分子间氢键的介绍
氢键是分子间相互作用的强健。向列相和近晶相稳定能量约是1千卡/摩 (多数柱状相是几千卡/摩)。氢键的强度大约是1-9千卡/摩,其中大部分是2-3千卡/摩。多数情况下,氢键的终端有一个棒状核心单位和氢键分子长轴方向平行5) ,在这种情况下,氢键不会抑制分子的旋转和液晶的稳定。然而,在分子短轴方向上的分子间的作用抑制分子的转动。
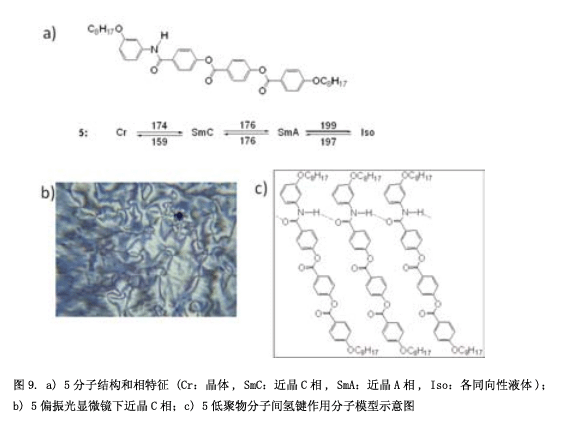
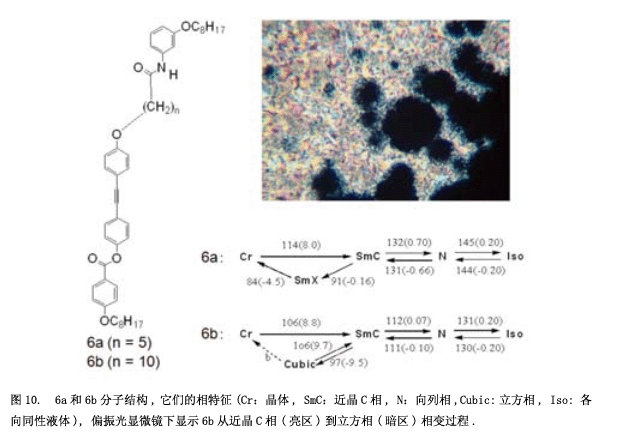
我们合成了一个具有横向方向氢键的化合物5(图9a),并研究其性质和分子堆积结构。在液晶相中,通过红外光谱图可以观测到氢键6) 。液晶相的温度范围非常窄,特别是近晶相C更窄(图9b)。假设是氢键阻碍了分子的转动,液晶相变得不稳定(图9c)。然后, 我们合成化合物6 (图10)具有一个“锚结构”的强的氢键和一个“螺旋结构”持续转动7) 。正与我们所料,化合物6显示出稳定的向列相和近晶相C。此外,也可以观测到一个立方相(图10中照片)。令人惊讶的是,这个立方相显示出自发的手性诱导作用,尽管这个分子并没有手性。自发的手性诱导在晶相中经常能够观测到,但是一般情况下,这种现象在液体或者是液晶流体中出现。可以假设这种现象通过刚性分子间氢键构建起来。从我们之前的结果来看,也可以认为这种自发的手性还原源于酯基的扭曲构型8) 。尽管分子没有任何手性,但是酯基(-CO-O-)在C-O单键和O键之间有一个左键或是右键,在第一阶段, 具有左键和右键的分子生成1:1的比例,然后又同样手性的分子组在一起成为一个分子聚集体。这种感应手性固定在每一个领域。此外, 我也计划了将一个很强的氢键脲引入分子(图11a)9) 。
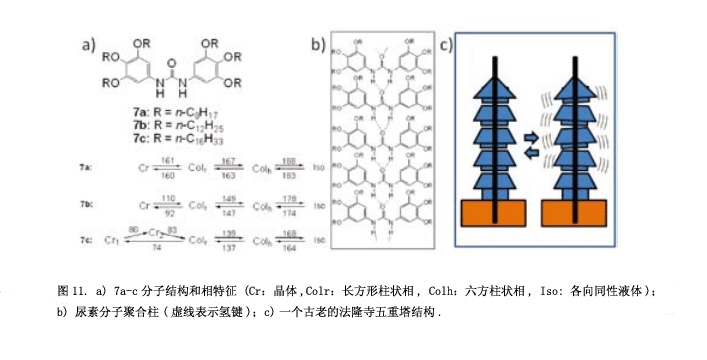
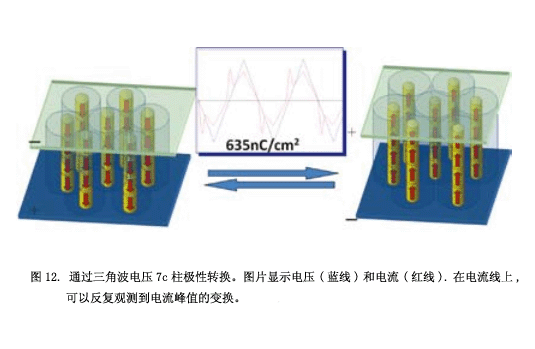
这个脲分子自行组成一个线性分子聚集体,如图11b所示。化合物7a-c在它们分子末端有六个大的烷基链的,分子间的空间位阻斥力在相邻的分子-NH-CO-NH-面之间产生了一个扭转角,从而形成一个螺旋的柱状结构。只有柱中心的部分有极性尿素分子,柱周边的部分是没有极性的烷基链。通常情况下,相邻的极性柱有一个反-平行的排列可以取消她们的极性和高稳定性而不改变液晶相的极性方向。然而, 在7a-c的结构中,柱状间的偶极相互作用远远小于其他液晶化合物柱,因为偶极间柱结构间的的长距。另外,中央线性尿素分子的重复距离是4.7Å,它们的苯环因为太长距离4.7Å,而没有强的分子间相互作用。这种结构类似于一个古老的法隆寺五重塔(图11c),这是世界上最古老的木结构建筑。所有的楼层都由贯穿中心宝塔的一个长而大的支柱支撑。当地震发生时,这座塔不会倒塌因为中心柔软的木质支柱。我们的尿素分子中心柱也有一个强大的线性氢键网结构,而分子的其他部分在相邻的分之间没有强的分子间作用,因此这个中心柱结构柔软并且晃动。因此,这写极性分子在灵活的极性柱配合下将他们的偶极子从一个方向转变到另一个方向,因此柱结构的极性可以改变方向(图12)。这个图片显示了当前的变化,并且应用一个三角波电压和合成7c液晶相中的细胞电容器。每一个分子的定向变化源于尿素分子的构象变化。目前为止,有几篇关于柱状相手性分子极性切换的报告10),但是没有柱状相非手性分子极性切换的例子11) 。我们关于这个转换性能的报道后,一些科学家声称转换峰源于离子杂质的运动。后来,柱极性转换通过二次谐波的产生实验证明12) 。
现在,我们正试图通过增加柱状间的距离或是增加转换能源缺口使得极性状态稳定。在不久的将来,我们将会按列控制柱的极性方向来实现高密度的存储装置。
4. 横向方向上全氟芳烃-芳烃间相互作用的介绍
1960年, Patrick和Prossor发现了六氟苯(mp 5.0 ℃) 和苯室温下(mp 5.4 ℃) 按1:1比例配合得到结晶 (mp 23.7 ℃),这个发现之后开创了全氟芳烃-芳烃间相互作用的历史13)。这个影响由范德华力和静电相互作用而成,能量是3.7-5.6千卡/摩14),这几乎与其中的氢键相同水平。据我们所知,通过全氟芳烃-芳烃间相互作用得到稳定的液晶相,只有两个例子是众所周知的15) 。在我们的实验室里,简单的化合物8具有一个全氟苯基和一个三烷氧基苯基(图13a) 16) 。
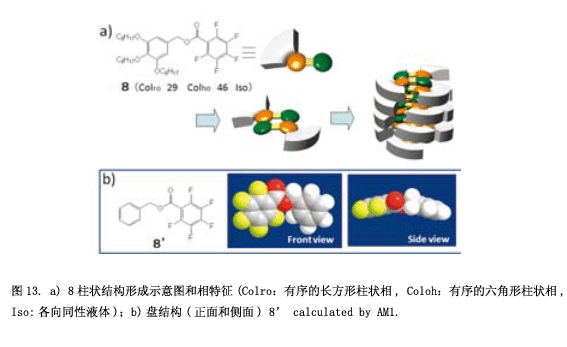
 如我们所料,这些分子在不同的π-面产生强烈的全氟芳烃-芳烃间相互作用,表现出柱状相。因为真空中分子8’(图13b)弯曲的杆状结构,产生了一个我们预计的宏观意义上可见的极性柱。然而,运用三角波电压下我们并没有观测到极性现象,这可能是因为分子间强的全氟芳烃-芳烃间相互作用的抑制。也有可能,每一个分子的柱结构与其相邻的分子中心部分(如图7所示的柱状结构)有极性转换的必要。在聚甲醛中,显示出很强的分子间作用,并且可以观测到磁带装纹理(图14a)。从二维XRD (图14b)可以看到,堆积在柱周边的分子组成了磁盘。
如我们所料,这些分子在不同的π-面产生强烈的全氟芳烃-芳烃间相互作用,表现出柱状相。因为真空中分子8’(图13b)弯曲的杆状结构,产生了一个我们预计的宏观意义上可见的极性柱。然而,运用三角波电压下我们并没有观测到极性现象,这可能是因为分子间强的全氟芳烃-芳烃间相互作用的抑制。也有可能,每一个分子的柱结构与其相邻的分子中心部分(如图7所示的柱状结构)有极性转换的必要。在聚甲醛中,显示出很强的分子间作用,并且可以观测到磁带装纹理(图14a)。从二维XRD (图14b)可以看到,堆积在柱周边的分子组成了磁盘。
5. 聚合近晶相分子模型的应用
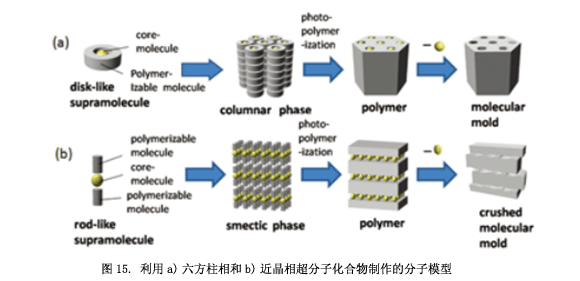
如图15a所示, 这是一个用于制造分子模具非常有用的六方柱状相17)。柱状构型的聚合在它们中心部分具有核心分子(或者离子),然后去除中心分子(或者离子)得到纳米多孔材料。这种材料有一个刚性的蜂窝状结构,对分子识别非常有用。另一方面,利用近晶相制作分子模型非常困难(图15b),从相应的聚合物中去除核心分子会导致层结构受到挤压。
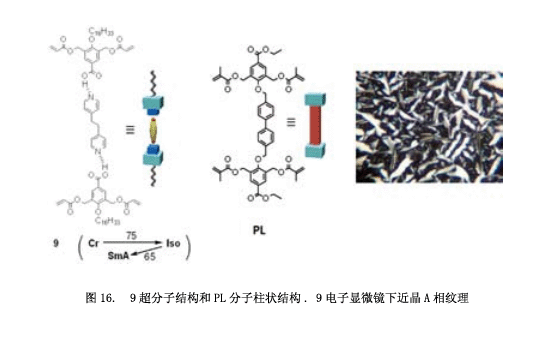
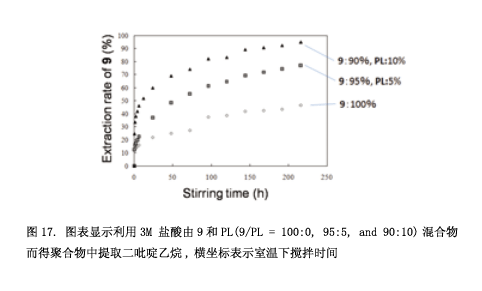
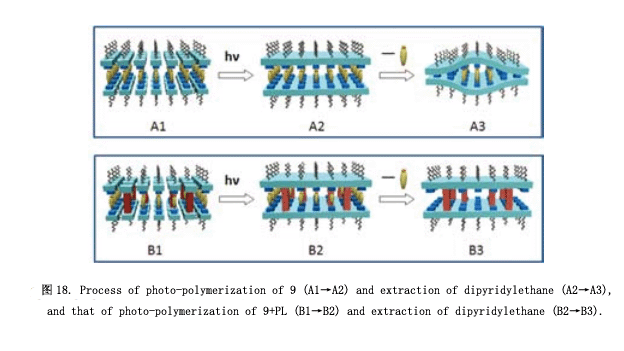
尽管如此,我们仍试图用近晶相合成分子模型18) 。如图16所示,超分子9 有一个二吡啶乙烷基分子作为核心分子,每一个终端通过氢键连接着一个p-烷氧基苯酸衍生物。两个聚合取代基横向引入到这个本算衍生物里面。我们也合成了一个杆状化合物PL ( “pillar”的缩写) ,其中的分子部分共价连接。化合物9展现出近晶相A (如图16微照片) ,这些分子里面有一个多层次的超晶格结构,纯化合物(9: 100%)光聚合得到相应的聚合物。然而,如图17所示,利用3M的盐酸从该聚合物中提取二吡啶乙烷非常缓慢。即使是216小时之后,仍有50%的二吡啶乙烷留在聚合物中。假设因为聚合物边缘层结构受到挤压使得这个核心分子不能够出去(图18: A1→A2→A3).然后,我们尝试着将PL分子引入分子层。将5% 的化合物9和10% 化合物PL混合得到相应的混合物,混合物光聚合为液晶相。从这个聚合物中提取核心分子,可以分别获得75% 和100% 核心分子。因此,这证实了近晶相超晶格结构通过引入PL可以用于制作分子模型。我们将要合成一种聚合物,通过引入PL分子与客体分子连接一起使得它具有一种很高的分子识别能力。
6.总结
从我得到一张写有关于液晶分子的文章已经12年过去了。我一直都在研究具有强烈横向互动的液晶分子。在分子的短轴方向上引入一个强大的作用力可以强烈的破坏液晶态的稳定,并且在多数情况下不会显示任何的液晶相。然而,如果我解决这些问题,我可以有很多机会来找到新的现象。并且我也可以极大地感觉到一种成就感。这些年,我有一个策略就是“在我的项目中,用最简单的分子可能来完成目的”,优点之一是,简单的化合物很容易得到,更为显著的优点是以简单分子合成目的会给许多科学家以强烈的冲击,并且他们中会有很多项目研究用到我们的分子。更进一步说,我们可以很容易的用简单的分子来解释一个新颖的现象,并且可以很容易的想象出它的应用。所以,我一直致力于研究变异的和简单的液晶分子的合成并从中找到有用的分子。
相关文献
1) K. Kishikawa, N. Muramatsu, S. Kohmoto, K. Yamaguchi, M. Yamamoto, Chem. Mater. 2003, 15, 3443.
2) K. Kishikawa, M. C. Harris, T. M. Swager, Chem. Mater.1999, 11, 867; S. H. Eichhorn, A. Paraskos, K. Kishikawa, T. M. Swager, J. Am. Chem. Soc. 2002, 124, 12742; I. A. Levitsky, K. Kishikawa, S. H. Eichhorn, T. M. Swager, J. Am. Chem. Soc. 2000, 122, 2474.
3) K. Kishikawa, Y. Miwa, T. Kurosaki, S. Kohmoto, M.Yamamoto, K. Yamaguchi, Chem. Mater. 2001, 13, 2468; T. Kajitani, Y. Miwa, N. Igawa, M. Katoh, S. Kohmoto, M. Yamamoto, K. Yamaguchi, K. Kishikawa, J. Mater. Chem.2004, 14, 2612.
4) K. Kishikawa, S. Furusawa, T. Yamaki, S. Kohmoto, M.Yamamoto, K. Yamaguchi, J. Am. Chem. Soc. 2002, 124,1597.
5) Reviews: T. Kato, N. Mizoshita, K. Kishimoto, Angew.Chem. Int. Ed. 2006, 45, 38; T. Kato, T. Yasuda, Y. Kamikawa, M. Yoshio, Chem. Commun. 2009, 729-739.
6) T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, J.Mater. Chem. 2004, 14, 3449; T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, Chem. Mater. 2004, 16, 2329; T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, Mol. Cryst. Liq. Cryst. 2005, 439, 2039.
7) T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa,Chem. Mater. 2005, 17, 3812.
8) T. Kajitani, H. Masu, S. Kohmoto, M. Yamamoto, K.Yamaguchi, K. Kishikawa, J. Am. Chem. Soc. 2005, 127,1124; S.-W. Choi, K. Fukuda, S. Nakahara, K. Kishikawa, Y. Takanishi, K. Ishikawa, J. Watanabe, H.Takezoe, Chem. Lett. 2006, 35, 896; S. Kawauchi, S.-W. Choi, K. Fukuda, K. Kishikawa, J. Watanabe, H. Takezoe, Chem. Lett. 2007, 36,750.
9) K. Kishikawa, S. Nakahara, Y. Nishikawa, S. Kohmoto, M. Yamamoto, J. Am. Chem. Soc. 2005, 127, 2565; H. Takezoe, K. Kishikawa, E. Gorecka, J. Mater. Chem. 2006,16, 2412.
10) H. Bock, W. Helfrich, Liq. Cryst. 1992, 12, 697; H. Bock, W. Helfrich, Liq. Cryst. 1995, 18, 387; H. Bock, W. Helfrich, Liq. Cryst. 1995, 18, 707; G. Heppke, D. Krüerke, M. Müller, H. Bock, Ferroelectrics 1996, 179, 203; G. Scherowsky, X. H. Chen, Liq. Cryst. 1994, 17, 803; G. Scherowsky, X. H. Chen, J. Mater. Chem. 1995, 5, 417; J. Barbera, R. Iglesias, J. L. Serrano, T. Sierra, M. R. de la Fuente, B. Palachios, M. A. Perez-Jubindo, J. T. Vazquez, J. Am. Chem. Soc. 1998, 120, 2908.
11) L. Lei, Mol. Cryst. Liq. Cryst. 1983, 91, 77; L. Lei, Mol. Cryst. Liq. Cryst. 1987, 146, 41; A. M. Levelut, J. Malthete, A. Collect. J. Phys. (France) 1986, 47, 351; H. Zimmerman, R. Poupko, Z. Luz, J. Billard, Z. Naturforsch., Teil A 1985, 1794; B. Xu, P. J. Carroll, T. M. Swager, Angew. Chem. Int. Ed. 1996, 35, 2094; M. Sawamura, K. Kawai, Y. Matsuo, K. Kanie, T. Kato, E. Nakamura, Nature
2002, 419, 702.
12) Y. Okada, S. Matsumoto, Y. Takanishi, K. Ishikawa, S.Nakahara, K. Kishikawa, H. Takezoe, Phys. Rev. E 2005,72, 020701(R); Y. Okada, S. Matsumoto, F. Araoka, M. Goto, Y. Takanishi, K. Ishikawa, S. Nakahara, K. Kishikawa, H. Takezoe, Phys. Rev. E 2007, 76, 041701.
13) C. R. Patrick, G. S. Prosser, Nature 1960, 187, 1021.
14) O. R. Lozman, R. J. Bushby, J. G. Vinter, J. Chem. Soc.Perkin Trans. 2 2001, 1446; S. Lorenzo, G. R. Lewis, I. Dance, New J. Chem. 2000, 24, 295; J. HernIndez-Trujillo, F. Colmenares, G. Cuevas, M. Costas, Chem. Phys. Lett. 1997, 265, 503; A. P. West, Jr., S. Mecozzi, D. A. Dougherty, J. Phys. Org. Chem. 1997, 10, 347.
15) M. Weck, A. L. Dunn, K. Matsumoto, G. W. Coates, E. B.Lobkovsky, R. H. Grubbs, Angew. Chem. Int. Ed. 1999,38, 2741; C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W.Clegg, C. Viney, Chem. Commun. 1999, 2493.
16) K. Kishikawa, K. Oda, S. Aikyo, S. Kohmoto, Angew.Chem. Int. Ed. 2007, 46, 764.
17) H.-K. Lee, H. Lee, Y. H. Ko, Y. J. Chang, N.-K. Oh, W.-C.Zin, K. Kim, Angew. Chem. Int. Ed. 2001, 40, 2669.
18) K. Kishikawa, A. Hirai, S. Kohmoto, Chem. Mater. 2008,20, 1931.
注:本文为提供者翻译的,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号