含较重主族元素的多重键化合物的新化学进展
- 2012-04-18
- 专题
1. 引言
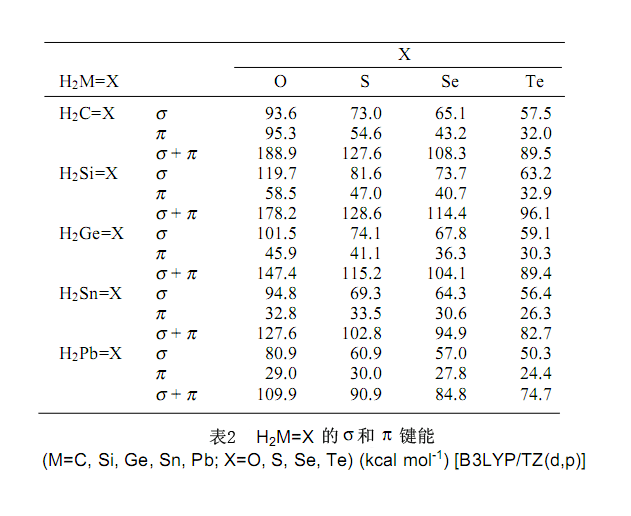
像烯烃、亚胺(希夫碱)、酮、乙炔、腈等含有第二周期元素的多重键化合物是稳定的化合物,并且在有机化学中扮演一个很重要的角色。与此相对的是,含有来自第三周期前面的元素(被称为较重的主族元素)的多键化合物有长键距,而且通过P-轨道重迭产生的π键能很小所以它们极不稳定。在表1中列举了π键能的一些例子。
因为这种不稳定性,直到1960年,所谓的“双键规则”在课本被描述为“含有来自第三周期前面的元素的稳定的双键是不存在的。”
然而,证实这些化合物存在于低温基质或气相中的证据已经从研究累积起来,这些研究开始于1960s后半叶到1970s之间。第一次合成并分离出含有P=C键3和Si=C4、Si=Si5、P=P6的稳定化合物分别在1978年和1981年。从那时起,关于与较重的主族元素相关的多重键化合物的化学发展迅速,并且几乎成为主族元素化学中的领导主题。在本文,我们介绍这个领域的最新进展,重点放在作者的研究上。
2. 含14族元素(硅,锗,锡,铅)的双键化合物
2.1 较重主族元素的烯烃类似物
2.1.1 双硅(Si-Si双键化合物)
以乙烯为代表的C-C双键化合物(烯烃)在有机化学中扮演着一个重要的角色。尽管烯烃有中等的反应活性,但烯烃通常是一个稳定的化合物。因此,很自然地,我们想尝试合成这种含有硅——同属于14族但位于碳下方——的双键化合物。然而,自从二十世纪末以来,这种实验结果失败了。获得的产品已经证实是它的低聚物或聚合物双键,而非双键化合物。在这种背景下,基于这些实验,前面提到的“双键规则”就成立了。已经被证实的是,含双键的碳和硅上位需取代基的引入作为一种防止聚合的手段是可行的。鉴于它通过降低与不稳定化学物质相关的反应速率来稳定,这种方法或将被命名为动力学稳定,或者因为它利用体积大的取代基保护高反应活性的双键而被命名为位保护。应用该技术,在1981年,Brook等4和West等5分别报道了含有第一个稳定碳-硅双键的硅烯1(AD=1-金刚烷基)和有一个硅-硅双键的二硅烯2(Mes=苯基)。在West报道的不久后,Masamune等用一种不同的方法合成了二硅烯3。8随后,又发展了几种制备硅烯9和二硅烯7的合成方法,所以现在知道了大量这类稳定的化合物。
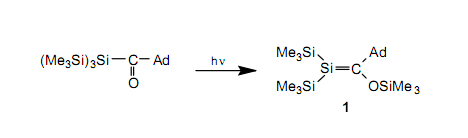
硅烯1,二硅烯2和二硅烯3在惰性的环境下是稳定的,但与图1中指出的烯烃相比,因为二硅烯有高HOMO和较低的LUMO,所以它们显示了极高的反应活性。相对于无色的烯烃,这是二硅烯是黄色的原因。
二硅烯2以方式4与氧气反应生成5。10进一步地,它与水、醇、酸等经历副反应后很容易生成6。11
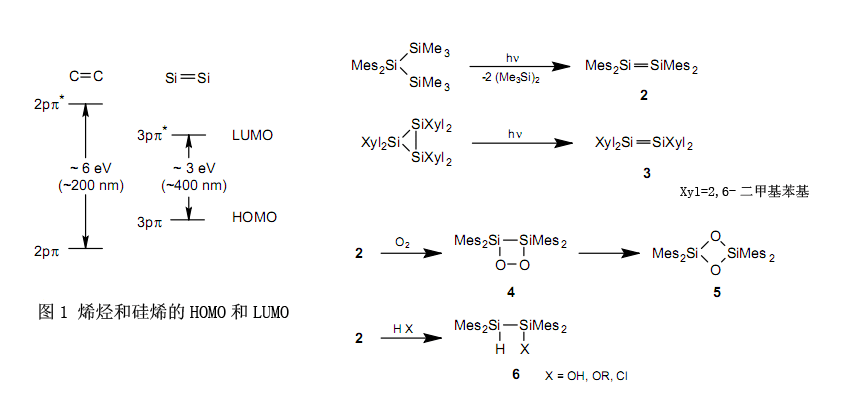
尽管事实上通过其他方法很难合成它们,但是利用二硅烯的高反应活性可以合成多种含有新结构的硅化合物。例如,通过与氧、硫、硒、碲、重氮甲烷、腈、叠氮化物等反应,以及与像苯乙酮这样的酮类反应,可以很容易的分别合成下述含硅的三环化合物712和四环化合物8。13

二硅烯2和3与空气接触时立刻分解。通过添加苯基到硅上,可以增加它们的稳定性。例如,由于大的金刚烷基和三异丙基苯基的存在,室温下化合物9和10的半衰期接近一天。我们已经能够合成比较稳定的化合物11,它室温下的半衰期大约一个月,这使它能在空气中使用。这种化合物有体积大的2,4,6-三[二(三甲基甲硅烷基)甲基]苯基基团(Tbt)。Tbt基团在邻位有四个体积大的三甲基甲硅烷基基团。然而,由于氢取代苄位,在1位有足够的空间,并且在这个方位上它允许发生足够的反应以进行功能基团转换。正如已被证实的,Tbt基团对稳定高周期的多键是非常有用的,这些多键通常有很高的反应活性。
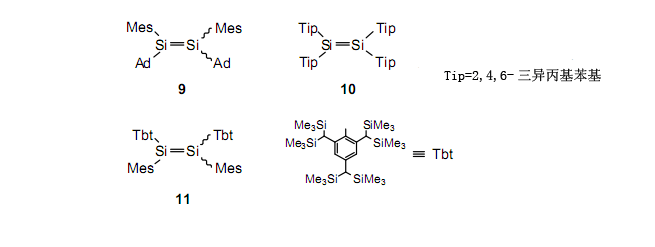
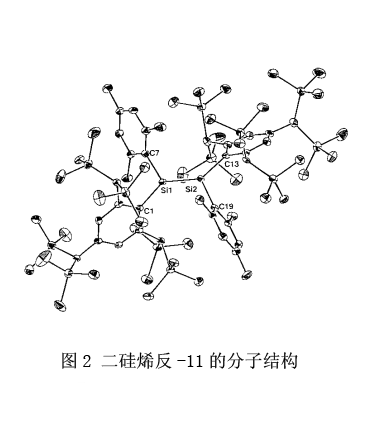
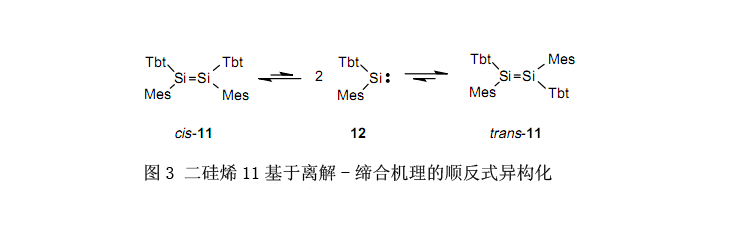
有趣的是,二硅烯11有极大的Tbt基团,所以能够在三个方向上保护Si-Si双键。另一方面,与其它二硅烯相比,硅-硅键被拉长所以很容易分解成硅烯。16 表216列举了二硅烯11(反式)的X射线晶体结构分析。在含有大的芳基取代基的二硅烯中最长的Si-Si键长是2.228Å。
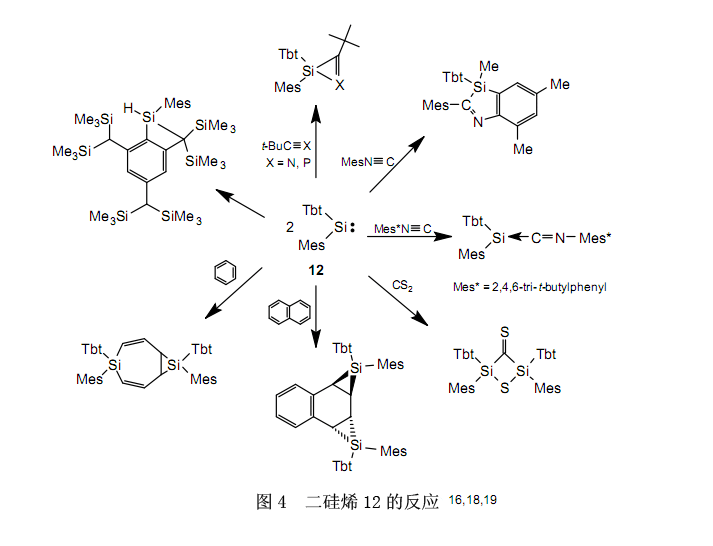
在溶液中,二硅烯11在室温下逐步分解成硅烯,而在70℃下极快分解。因此,在室温下经由硅烯的分解,反式-二硅烯11异构化为顺式-二硅烯11和顺式-二硅烯11异构化为反式-二硅烯11分别进行。众所周知,烯烃的异构化作用不是通过C-C键的裂解发生, 而是通过键的旋转。而且,假设在硅上含有相对较小尺寸的取代基的已知二硅烯,异构化作用与在烯烃上一样通过Si-Si键的旋转产生。二硅烯11是第一个基于离解-缔合规则异构化的二硅烯,了解这一点是很有趣的(图3)。虽然至今为止通过热解或光解作用来生产硅烯,但是在温和的环境下产生硅烯方法的发现已经使合成新的有机硅化合物成为可能,尽管事实上难以利用以前的方法合成它。图4列举了几个例子。16,18,19
2.1.2 锗、锡、铅双键化合物(二锗烯、锡烯、铅烯)
因为关于Ge=Ge、Sn=Sn、Pb=Pb的π键能相对于涉及Si=Si的π键能比较小,所以含有与硅属于同一族的锗、锡、铅的双键化合物(命名为二锗烯、锡烯、铅烯)也同样比较不稳定。另一方面,经由谈及的双键分解产生的二价化学物种锗烯、锡烯和铅烯是非常稳定的。因此,有适当尺寸的取代基对于稳定的双键化合物是很重要的。
虽然在1970s,Lappert等在二硅烯2未合成时先合成了二锗烯1321,22和锡烯1421,22,但是研究Ge=Ge和Sn=Sn的性质是不可能的,因为他们以锗烯15和stannylene16的形式分别存在于溶液中,尽管事实上它们作为双键化合物存在于固相。二硅烯2和3合成之后,进行了不引起分解而合成二锗烯和锡烯的研究。结果是合成了化合物1723和1824。
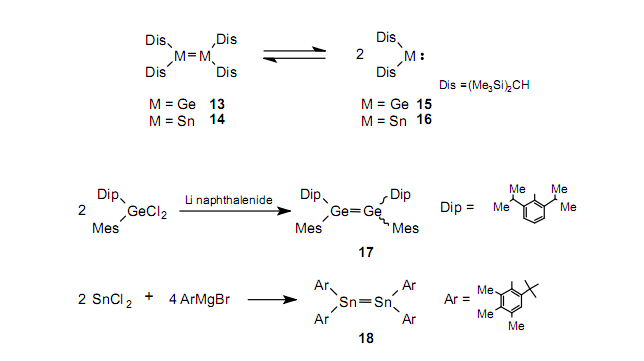
虽然二锗烯17不分解成锗烯,但是含有较大取代基的二锗烯19易分解成锗烯20,并在两种物质间存在一个平衡。25
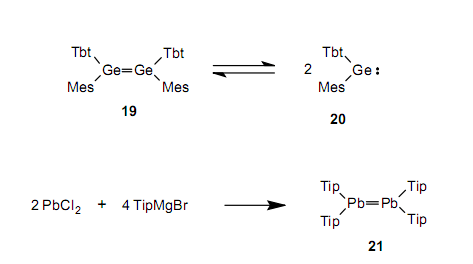
在1999年,也合成了含一个双键的铅烯21,这个双键在占据周期表中最低位置的铅原子之间。26因此,在west等合成二硅烯2后的18年里,已经完成了属于14族的相同元素之间的稳定的双键化合物。
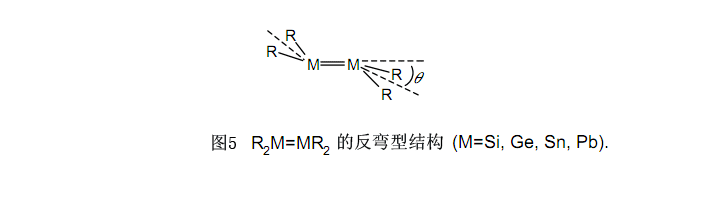
例如2,17-19,21的R2M=MR2化合物(M=Si,Ge,Sn,Pb)具有有趣的结构特性,这是不同于烯烃的。这些化合物有一个所谓的“反弯型结构”。7,20如在图5中所列举的,在一个M上的两个取代基和另一个M上的两个取代基以q被分弯曲成反式,而当原子数增加时与平面形成的弯曲角将变大。虽然在本文未描述细节,但是据透露,已经通过R2M:的单线态和三线态间的能量差异确定了弯曲角,而且采取这样一个与单线态稳定程度相称的弯曲结构是比较恰当的。
2.2 含硅的芳香族化合物
和烯烃一样,含有不饱和键的主要有机化合物是包括苯,萘等在内的芳香族化合物。因此,硅取代苯和萘中的碳得到的化合物是否展现出芳香族的性质,这个问题引起了极大的兴趣。在1970s到1980s这个时期里进行了大量的研究。在低温基质(例如,10K下的氩基质)或在气相中,在硅上有像羟基,甲基基团等简单取代基的硅杂苯22(R=H,Me)可以存在一小段时间,而且已经测定了UV,IR光谱等。28与此同时,在1988年Märkl等报道的23已经是一个适度稳定的独立的化合物,尽管事实上为分离作为一个稳定化合物的硅杂苯已经做了大量的实验。29虽然硅上的一个异丁基基团和邻位的两个三甲基甲硅烷基基团位保护了23,但是它只是在-100℃下的溶液中稳定。此外,在后面会介绍,内环硅通过溶液中四氢呋喃的协调确保稳定。
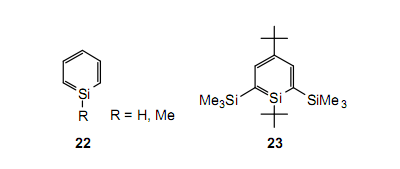
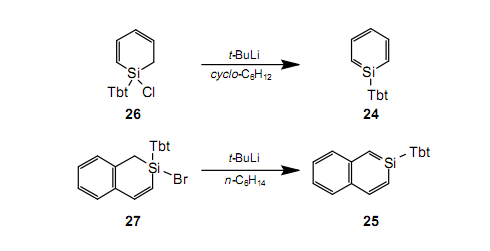
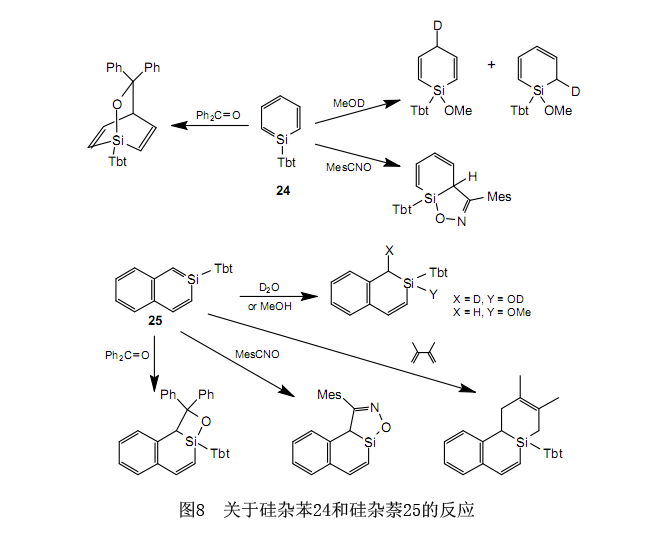
通过把Tbt基团出色的保护能力应用到含硅的芳香族化合物的合成上,我们已经成功地分别分离出在室温下作为稳定晶体的硅杂苯2430和硅杂萘2531。24和25的合成前驱体分别是卤化硅26和27。通过使用异丁基锂时前驱体的脱氢卤化作用获得产品。
24和25都是无色的晶体,它们在惰性气氛下非常稳定以至于在结晶态或溶液中完全不改变。29根据29Si的NMR,内环硅的化学位移分别是87.3和92.5,这种情况存在于sp2硅的低磁场区域特性中。这完全不同于Märkl等报道的化合物23的26.8的化学位移。在这种情况下,人们认为作为溶剂的四氢呋喃明显与23协调,所以相应地,它在较大程度上是稳定的。表6列举了内环硅的耦合常数。两个相邻原子间的耦合常数与涉及键合的键级相联系。依据图6,硅杂苯24的耦合常数在硅杂萘25的两个耦合常数之间,这恰好与苯和萘键级的关系是一致的。另外,一种典型的Si-C单键的耦合常数大约是50Hz。因此,24和25的Si-C键明显具有了一个双键的特征。结果是,π电子的离域作用明显存在于24和25中,而且它们具有芳香族的特性。
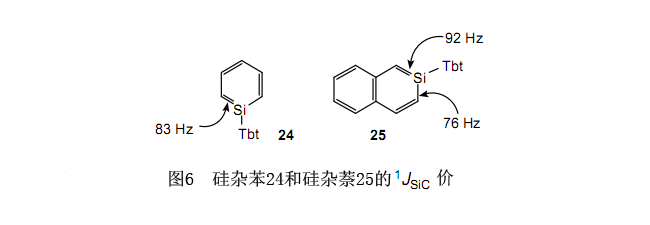
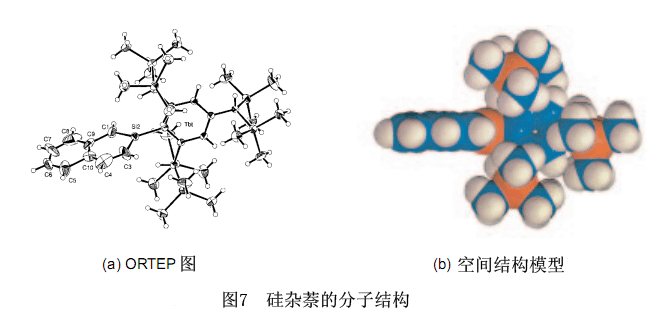
图7显示了25的X射线晶体结构分析。硅杂苯的环状部分位于平面上,而内环硅和结合在它上的三个原子位于同一平面上。进一步地,高活性的内环硅完全由Tbt基团保护,从空间结构模型(b)中可以清楚的看到。
从根本上说,24和25的电子光谱彼此相似,尽管事实上它们转移到比相应的苯和萘更长的波长。这也支持了24和25有芳香族特性这种情况。同时,芳香族特性的量子计算理论变得更为可靠。Schleyer等建议NICS(核独立化学位移)是一个关于芳香族特性的良好的指标。32作为涉及硅杂苯与1-硅杂萘,2-硅杂萘和9-硅杂萘进行的NICS计算的结果,他们的芳香族特性几乎相同,尽管事实上它们比苯和萘的稍低,而且更进一步,关于芳香族特性,硅杂萘的三种异构体之间几乎没有差别。31计算的结果与从NMR,UV,X射线晶体结构分析等中得到的实验结果是一致的。24和25的活性是很高的,尽管事实上它们在硅上有体积大的取代基并有推测的芳香族特性。在图8中列举了它们反应的几个例子。我们推断,这是以与Si=C双键相关的高反应活性超过了来自于牵涉到硅杂苯和硅杂萘芳香族特性的稳定性为依据的。
3. 含15族元素的双键化合物(磷,砷,锑和铋)
含N-N双键的偶氮化合物也是与烯烃一样众所周知的化合物。类似地,在合成出硅烯1和二硅烯26同一年的1981年,yoshifuji等也第一次合成了含有一个稳定P-P双键的化合物——二磷烯28。
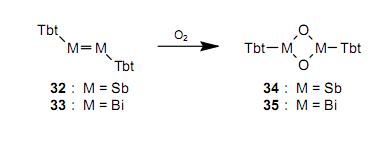
随即,使用一个简单的方法也合成了一个含有As-As双键的稳定的化合物二砷烯。34由于这些双键是逐渐不稳定,所以尽管进行了很多实验,但是从未分别合成含有Sb-Sb和Bi-Bi之间双键的化合物二锑烯和二铋烯。人们期望,即使对15族中元素之间的双键化合物, Tbt基团也能够提供位保护。在研究了很多合成方法之后,我们已经第一时间分别合成并分离出作为绿色和紫色晶体的稳定的二锑烯3235和二铋烯3336。我们发明了一种新的合成方法,它通过使用三价磷试剂从六元化合物30和31上除去硒。
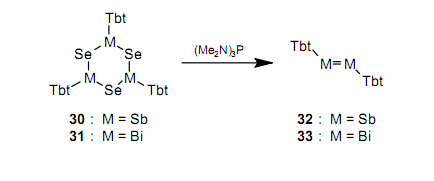
在有稳定同位素的元素中,铋是有最大的原子序数的第六周期元素。关于二铋烯33的合成,人们的浓厚兴趣不仅来自即使第六周期元素都可能产生双键这一观点(如前所述,在33合成之后,也合成了作为一个在其他的第六周期元素之间的双键化合物二铅烯21),而且也是来自它是有最大原子量的元素的双键化合物的合成。通过X射线晶体结构分析确定了与32和33有关的结构。图9列举了33的分子结构,36从这里我们能够推断出体积大的Tbt基团有效地保护了有高反应活性的Bi-Bi双键。
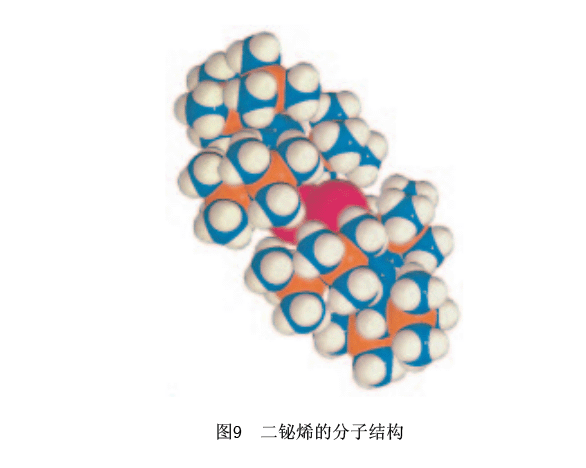
处于晶体态的32和33 是相当稳定的化合物,所以在空气中,我们可以在短期内处理它们。然而在空气中,它们逐步与氧气反应产生四元环化合物34和35。非常有趣的是反应通过维持晶相进行,并且产生了单晶体34和35。35,36通过使用快速X射线衍射设备追踪32的反应,可以发现反应在大约20小时的诱导期之后突然开始。据推测,在氧气达到了一个临界浓度后,氧气从晶体表面进入32,而且氧气和32之间的反应在一个像倒下的多米诺骨牌的拉伸下进行。非常有趣的是当维持单一晶相时没有这种来自外部的反应物反应的例子。
4. 含16族元素(硫、硒、碲)的双键化合物
不夸张地说,在有机化学,特别是有机合成化学中,由第二周期元素产生的最重要的双键化合物是以醛,酮等为代表的羰基化合物。研究一种正在讨论中,以高周期元素取代羰基中氧原子的作用是极为有趣的。
4.1 碲酮
虽然很久之前已经知道硫取代氧的硫酮(R2C=S),但是硒酮(R2C=S)是在1970s中期被Barton等合成出来。37Barton等并不能合成碲酮(R2C=Te)。
我们已经开发出腙和二氯二硫38或二氯二硒39的反应,作为一种硫烯和硒烯的新合成方法。

通过把上述的反应应用到二氯化碲或四氯化碲,40a我们得到了含有杂环telluradiazoline36,通过热解我们能够由它合成第一个稳定的碲酮37。40b

4.2 酮的较重元素类似物(重酮)
如果由来自14族较高周期的元素,例如硅、锗、锡或铅,取代酮中碳原子,产生的化合物就是我们指的当作重酮的酮的较重元素类似物。此外,如果由来自16族的其他元素,例如硫、硒或碲取代氧原子,得到的化合物也会被指为重酮。
就像表2指出的,碳氧双键的σ和π键能大约相当。另一方面,所有的重酮中,σ键是强于π键的。因此,当酮易于进行加成和消去反应时,重酮更易于进行由两个更积极稳定的σ键取代一个π键的加成反应。由于这些积极地原因,合成重酮是很困难的。我们已经能够合成重酮化合物,这种物质中体积大的取代基减少了π键的反应活性。
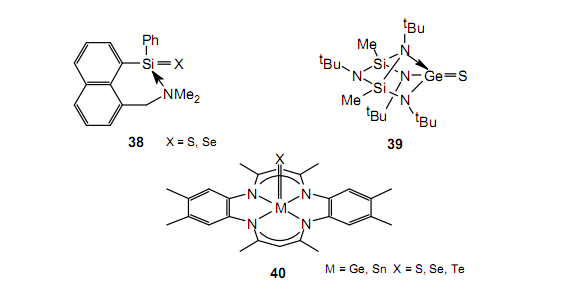
为了合成与重酮相关的双重键化合物已经进行了很多合成实验。在化合物38-40中已经列举了几个例子。其中一些并不是真的双键化合物,因为在某种程度上通过杂原子的配位稳定了双键的特征。
这一次,我们也是通过使用高位保护能力的Tbt基团才能合成许多稳定的重酮。图10列举了两种使用的合成方法。总之,方法(1)比较常见,但是当作为开始原料的1,2,3,4-tetrachalcogenametalloranes的合成不是那么容易时,方法(2)将会有用。

当Tbt和Tip基团共同存在于14族元素上时,把硅烷硫酮4142、锗烷硫酮4246,47锗烷硒酮4347,48当作稳定的化合物分别分离出来是可以实现的。
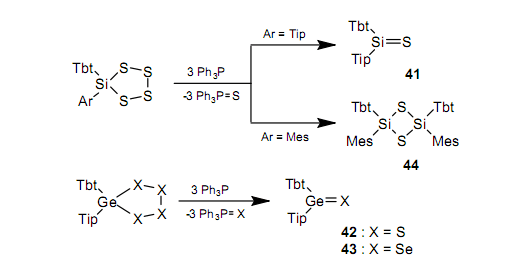
然而,当一个较小的Mes基团取代了Tip时,分离的化合物是一个二聚物44,虽然我们能够确认通过被困反应产生了作为中间体的Tbt(Mes)Si=S。所以为了分离重酮,具有位保护的体积大的取代基是很重要的。42依据Tbt和Tip取代基化合的方法不足以合成含锡的重酮。这是由于当与含锡双键相关的能量降低时提升了反应活性,而Sn周围的双键变得更长时,位保护作用未有效地发挥作用。然而,在一个稳定相中,凭借把Tip改为一个较大的取代基Ditp基团,我们也能够分离锡烷硫酮4549和锡烷硒酮4650。
通过使用被困反应,在较低温度下,能够在脱硫作用中确认铅烷硫酮47,但是不可能分离出47。
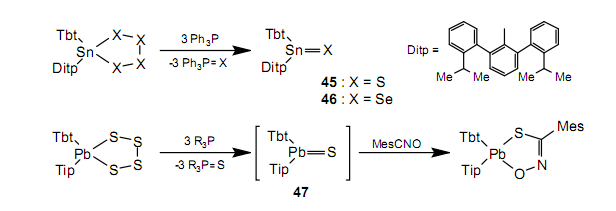
我们不能合成出相应的tetraselenasilorane(R1R2SiSe4),所以我们无法通过脱硒化合成silaneselone(R1R2Si=Se)。然而通过使用silacyclopropane48作为硅烯49的先驱体的硒化反应,我们可以合成silaneselone50。52
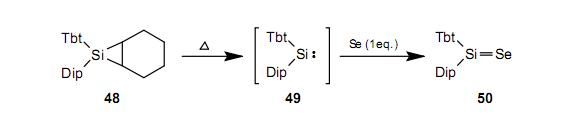
至于锗,锗环丙烯51,52起到作锗烯 53,5453良好的先驱体。通过在硫和硒存在时加热52,我们能够获得锗烷硫酮55和锗烷硒酮56。47对于Ge-Te双键合成,这个反应是特别有效的,而且我们能够合出锗烷碲酮57,58。54
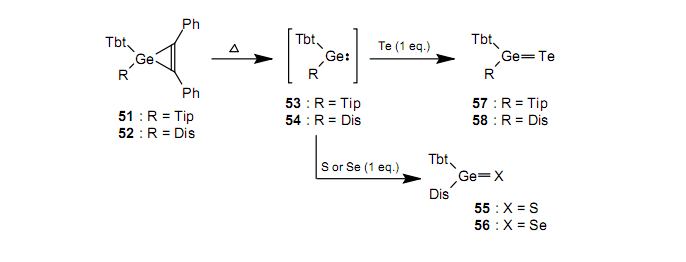
当我们把分离出的铅烯59与相当量的硫反应时,我们得到了一种二价的化学物种铅烯61。55
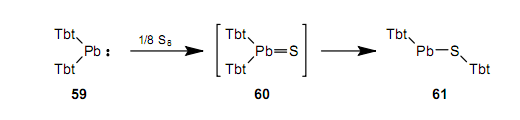
因此,就铅而言,2-配位型61比3-配位型60更稳定。这与量子化学计算的结果是一致的,结果显示HPb(SH)比H2Pb=S稳定了大约39kcal/mol。41a有关铅的重酮有不同于轻类似物的性质。
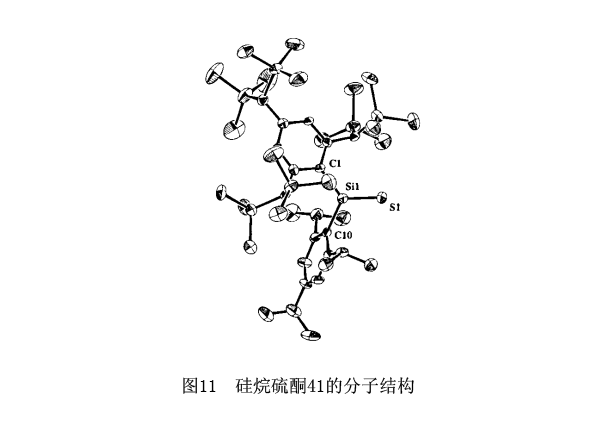
所有合成的重酮被观察成有色晶体(41:黄色;42,55,黄橙色;43,45,46,50,56:红色;57,58,绿色)。X射线晶体结构分析确定了硅烷硫酮41,锗烷硫酮42,锗烷硒酮43,56,锡烷硒酮46和锗烷碲酮57,58的结构。图11列举了硅烷硫酮41的分子结构。42
值得关注的结构特性是硅周围全部三角平面协调的键角都是360°,而且Si-S键距是1.948 Å,这比普通的Si-Si单键短到大约9%,所以很明显它有双键的特征。对于酮,所有这些数据都是完全相同的结构特性。已经得到实验证实的是重酮与酮结构上是相同的。X射线晶体结构分析确定的其他重酮呈现一样的结构特性。
在碳-氧双键的反应中,关于羰基基团的重要反应特性是可逆性,如在与水和醇的反应中所示的一样。在讨论中,基于加成-消去机理的反应经由一个四配位的中间物进行。
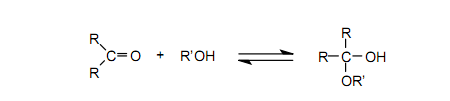
在表2中看到,与C=O键相关,大约相等的σ键能和π键能使前面提到的方法成为可能。然而,假设重酮,加成反应是显著放热并且不可逆,其原因很大程度上是π键能比σ键能低。事实上,所有合成的重酮迅速与水、醇等反应并引发四配位副产物。另外,相对于酮的情况,重酮很容易和不饱和的化学物,例如苯基异硫氰酸盐、mesitonitrile oxide、2,3-二甲基丁二烯等发生环加成反应。42-50通过使用这些反应我们能够合成新的杂环化合物,尽管事实上通过一种其他方法难以合成它们。

5. 结论
通过使用体积大的取代基,在动力学稳定性基础上合成了大量包含14,15和16族高周期元素的多重键化合物。更进一步地,虽然我们在本文中没有涉及它们,但是也已经合成了一些包含13族元素的多重键化合物。与高周期元素有关的多重键化合物与含有有趣结构和反应活性的独特化合物一起已经在有机化学和无机化学中建立了一个新的领域。然而,在这个领域中仍然留有大量正等待合成的化合物。例如,在14族元素之间的三键化合物,R-M≡M-R,含氧的重酮,R2M=O(M=Si,Ge,Sn和Pb)以及含Ge,Sn,Pb等的芳香族化合物。
参考文献:
1) M. W. Schmidt, P. N. Truong, M. S. Gordon, J. Am. Chem. Soc., 109, 5217 (1987); P. v. R. Schleyer, D. Kost,J. Am. Chem. Soc., 110, 2105 (1988).
2) K. S. Pitzer, J. Am. Chem. Soc., 70, 2140 (1948).
3) T. C. Klebach, R. Lourens, F. Bickelhaupt, J. Am. Chem. Soc., 100, 4886 (1978).
4) A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst, R. K. Kallury, J. Chem. Soc., Chem. Commun.,1981, 191.
5) R. West, M. J. Fink, J. Michl, Science, 214, 1343 (1981).
6) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu, T. Higuchi, J. Am. Chem. Soc., 103, 4587 (1981).
7) Review: R. Okazaki, R. West, Adv. Organomet. Chem., 39, 233 (1996).
8) S. Masamune, Y. Hanzawa, S. Murakami, T. Bally, J. F. Blount, J. Am. Chem. Soc., 104, 1150 (1982).
9) Review: A. G. Brook, M. A. Brook, Adv. Organomet. Chem., 39, 71 (1996).
10) A. J. Millevolte, D. R. Powell, S. G. Johnson, R. West, Organometallics, 11, 1091 (1992); K. L. McKillop,G. R. Gillette, D. R. Powell, R. West, J. Am. Chem. Soc., 114, 5203 (1992); H. Sohn, R. P. Tan, D. R. Powell,R. West, Organometallics, 13, 1390 (1994).
11) D. N. Roark, G. J. D. Peddle, J. Am. Chem. Soc., 94, 5837 (1972); D. J. DeYoung, M. J. Fink, J. Michl,R. West, Main Group Met. Chem., 1, 19 (1987).
12) H. Piana, U. Schubert, J. Organomet. Chem., 348, C19 (1988); G. R. Gillette, R. West, J. Organomet. Chem.,394, 45 (1990); H. B. Yokelson, A. J. Millevolte, G. R. Gillette, R. West, J. Am. Chem. Soc., 109, 6865(1987); R. West, D. J. DeYoung, K. J. Haller, J. Am. Chem. Soc., 107, 4942 (1985); R. P. Tan, G. R. Gillette,D. R. Powell, R. West, Organometallics, 10, 546 (1991); H. B. Yokelson, A. J. Millevolte, K. J. Haller, R. West,J. Chem. Soc., Chem. Commun., 1987, 1605.
13) M. J. Fink, D. J. DeYoung, R. West, J, Michl, J. Am. Chem. Soc., 105, 1070 (1983); P. Boudjouk, B.-H. Han,K. R. Anderson, J. Am. Chem. Soc., 104, 4992 (1982); P. Boudjouk, Nachr. Chem., Tech. Lab., 31, 798(1983); A. D. Fanta, D. J. DeYoung, J. Belzner, R. West, Organometallics, 10, 3466 (1991).
14) B. D. Shepherd, D. R. Powell, R. West, Organometallics, 8, 2664 (1989).
15) R. S. Archibald, Y. van den Winkel, D. R. Powell, R. West, J. Organomet. Chem., 446, 67 (1993); H. Watanabe,K. Takeuchi, N. Fukawa, M. Kato, M. Goto, Y. Nagai, Chem. Lett., 1987, 1341.
16) N. Tokitoh, H. Suzuki, R. Okazaki, K. Ogawa, J. Am. Chem. Soc., 115, 10428 (1993); H. Suzuki, N. Tokitoh,R. Okazaki, J. Harada, K. Ogawa, S. Tomoda, M. Goto, Organometallics, 14, 1016 (1995); H. Suzuki,N. Tokitoh, R. Okazaki, Bull. Chem. Soc. Jpn., 68, 2471 (1995).
17) a) R. Okazaki, M. Unno, N. Inamoto, Chem. Lett., 1987, 2293. b) R. Okazaki, N. Tokitoh, T. Matsumoto,in “Synthetic Methods of Organometallic and Inorganic Chemistry,” ed by W. A. Herrmann, Thieme,New York (1996) Vol. 2, p. 260.
18) H. Suzuki, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 116, 11572 (1994).
19) N. Tokitoh, H. Suzuki, R. Okazaki, J. Chem. Soc., Chem. Commun., 1996, 125; N. Takeda, H. Suzuki,N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 119, 1456 (1997).
20) Review: K. Baines, W. G. Stibbs, Adv. Oranomet. Chem., 39, 275 (1996).
21) P. J. Davidson, D. H. Harris, M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268.
22) D. E. Goldberg, P. B. Hitchcock, M. F. Lappert, J. Chem. Soc., Dalton Trans., 1986, 2387.
23) S. A. Batcheller, T. Tsumuraya, O. Tempkin, W. M. Davis, S. Masamune, J. Am. Chem. Soc., 112, 9394(1990).
24) M. Weidenbruch, H. Killian, K. Peters, H. G. v. Schnering, Chem. Ber., 128, 983 (1995).
25) K. Kishikawa, N. Tokitoh, R. Okazaki, Chem. Lett., 1998, 239.
26) M. Stürmann, W. Saak, H. Marsmann, M. Weidenbruch, Angew. Chem., Int. Ed., 38, 187 (1999).
27) Y. Apeloig, M. Karni, in “The Chemistry of Organic Silicon Compounds,” ed by Z. Rappoport, Y. Apeloig,Wiley, New York (1998) Vol. 2, Chapter 1.
28) T. J. Barton, D. Banasink, J. Am. Chem. Soc., 99, 5199 (1977); H. Bock, R. A, Bowling, B. Solouki,T. J. Barton, G. T. Burns, J. Am. Chem. Soc., 102, 429 (1980); T. J. Barton, G. T. Burns, J. Am. Chem. Soc.,100, 5246 (1978); G. Maier, G. Mihm, H. P. Reisenauer, Angew. Chem., Int. Ed. Engl., 19, 52 (1980).
29) G. Märkl, W. Schlosser, Angew. Chem., Int. Ed. Engl., 27, 963 (1988).
30) K. Wakita, N. Tokitoh, R. Okazaki, S. Nagase, Angew. Chem., Int. Ed., 39, 634 (2000).
31) N. Tokitoh, K. Wakita, R. Okazaki, S. Nagase, P. v. R. Schleyer, H. Jiao, J. Am. Chem. Soc., 119, 6951 (1997);K. Wakita, N. Tokitoh, R. Okazaki, S. Nagase, P. v. R. Schleyer, H. Jiao, J. Am. Chem. Soc., 122, 5648 (2000).
32) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. v. E. Hommes, J. Am. Chem. Soc., 118, 6317(1996).
33) M. Yoshifuji, in “Multiple Bonds and Low Coordination in Phosphorus Chemistry,” ed by M. Regitz,O. J. Scherer, Thieme, New York (1990), Chapter 9.
34) A. Cowley, J. G. Lasck, N. C. Norman, M. Pakulski, J. Am. Chem. Soc., 105, 5506 (1983).
35) N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa, Y. Ohashi, J. Am. Chem. Soc., 120, 433(1998).
36) N. Tokitoh, Y. Arai, R. Okazaki, S. Nagase, Science, 277, 78 (1997).
37) T. G. Back, D. H. R. Barton, M. R. Britten-Kelly, F. S. Guziec, Jr., J. Chem. Soc., Perkin Trans. 1, 1976, 2079.
38) R. Okazaki, K. Inoue, N. Inamoto, Bull. Chem. Soc. Jpn., 54, 3541 (1981).
39) A. Ishii, R. Okazaki, N. Inamoto, Bull. Chem. Soc. Jpn., 61, 861 (1988).
40) a) M. Minoura, T. Kawashima, N. Tokitoh, R. Okazaki, Tetrahedron, 53, 8137 (1997). b) M. Minoura,T. Kawashima, R. Okazaki, J. Am. Chem. Soc., 115, 7019 (1993); M. Minoura, T. Kawashima, R. Okazaki,Tetrahedron Lett., 38, 2501 (1997).
41) Reviews: a) N. Kano, N. Tokitoh, R. Okazaki, Yuki Gosei Kagaku Kyokai Shi (J. Synth. Org. Chem. Japan),56, 919 (1998). b) N. Tokitoh, T. Matsumoto, R. Okazaki, Bull. Chem. Soc. Jpn., 72, 1665 (1999). c) N. Tokitoh, R. Okazaki, in “ The Chemistry of Organic Silicon Compounds,” ed by Z. Rappoport,Y. Apeloig, Wiley, New York (1998) Vol. 2, Chapter 17.
42) H. Suzuki, N. Tokitoh, R. Okazaki, S. Ngase, M. Goto, J. Am. Chem. Soc., 120, 11096 (1998).
43) R. Arya, J. Boyer, F. Carré, R. Corriu, G. Lanneau, J. Lapasset, M. Perrot, C. Priou, Angew. Chem., Int. Ed,Engl., 28, 1016 (1989).
44) a) M. Veith, S. Becker, V. Huch, Angew. Chem., Int. Ed, Engl., 28, 1237 (1989). b) M. Veith, A. Detemple,V. Huch, Chem. Ber., 124, 1135 (1991). c) M. Veith, A. Detemple, Phosphorus, Sulfur, Silicon, 65, 17 (1992).
45) a) M. C. Kuchta, G. Parkin, J. Chem. Soc., Chem. Commun., 1994, 1351. b) M. C. Kuchta, G. Parkin,J. Am. Chem. Soc., 116, 8372 (1994). c) W.-P. Leung, W.-H. Kwok, L. T. C. Law, Z.-Y. Zhou, T. C. W. Mak,J. Chem. Soc., Chem. Commun., 1996, 505.
46) N. Tokitoh, T. Matsumoto, K. Manmaru, R. Okazaki, J. Am. Chem. Soc., 115, 8855 (1993).
47) T. Matsumoto, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 121, 8811 (1999).
48) T. Matsumoto, N. Tokitoh, R. Okazaki, Angew. Chem., Int. Ed. Engl., 33, 2316 (1994).
49) M. Saito, Ph.D. Thesis, The University of Tokyo, 1996.
50) M. Saito, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 119, 11124 (1997).
51) N. Kano, N. Tokitoh, R. Okazaki, Chem. Lett., 1997, 277; N. Kano, N. Tokitoh, R. Okazaki, Organometallics,16, 4237 (1997).
52) T. Sadahiro, N. Tokitoh, R. Okazaki, unpublished results.
53) N. Tokitoh, K. Kishikawa, T. Matsumoto, R. Okazaki, Chem. Lett., 1995, 827.
54) N. Tokitoh, T. Matsumoto, R. Okazaki, J. Am. Chem. Soc., 119, 2337 (1997).
55) N. Kano, Ph.D. Thesis, The University of Tokyo, 1998.
注:本文为提供者翻译的,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号