碘资源的利用—高价有机碘的合成和反应
- 2012-04-19
- 专题
目前我们的研究重点是低毒性的三价超价有机碘(λ3—有机碘),以及其在有机化学合成中的充分利用。
碘是大原子半径的卤族元素,具有易极化和低负电性的特点。八隅体理论指出碘能够形成高价态有机碘是由于其易升高的价键所致。
典型的λ3—有机碘有亚酰碘苯(聚合物)1,碘苯二乙酸2,双(三氟乙酰氧基)碘苯3,羟基(对甲苯磺酰氧基)碘苯(科泽试剂)4,2—亚碘酰基苯甲酸5。它们作为氧化剂广泛应用于活性亚甲基、双键和三键、醇羟基和酚羟基、硫基和氨基化合物的氧化反应1)。包括上述常见的碘类物质,有20多种的λ3—有机碘化合物可直接从东京Kasei Kogyo公司购买。
由于过氧烷基基团的λ3—有机碘化合物具有较强的易分解性,因此目前还不能成功合成。我们最近研究发现2)路易斯酸催化使1-羟基-1,2苯碘酰基-3(1氢)-酮 5和过氧叔丁酸进行配位体交换,得到过氧碘化物6晶体。过氧碘化物6在固体状态下非常稳定,不易分解,而且它的结晶形态在室温下可以较好地保存一年以上。这个产品引起人们的关注是因为它在同一个分子中含有过氧化叔丁基和一个三价的碘原子,而两者都是极强的氧化剂。本文的目的是为了说明λ3—过氧碘化物的有用性及其作为自由基强氧化剂的广泛应用。
1.烯基碘化物和炔基碘化物的合成与反应
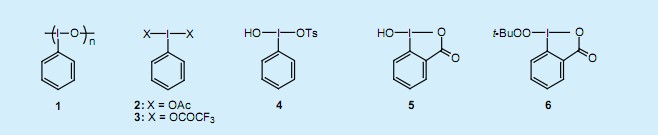
在1985年人们发现亚碘酰苯1与BF3在二氯甲烷中活化,并与乙烯基硅烷或乙烯基锡烷反应,其中硅或者锡被三价的碘取代,生成λ3—烯基-碘化合物7并能取得较好产率(图1)3)。λ3—烯基-碘化合物的生成是立体定向的,且乙烯基硅烷和乙烯基锡烷的立体化学结构保持不变。λ3—烯基-碘化合物7也可以通过以乙烯基硼酸为基质的碘化硼交换反应制得。
三价碘基团具有很强的基团离去性能,因此烯基碘化物7中乙烯基碳的亲核取代反应在室温的温和条件下就能进行。在这些反应中,烯醇基阴离子、R2CuLi、CuCN、ArSNa、n-Bu4NC等可作为良好的亲核试剂。特别的,伴随着立体化学构型的转化,发生了乙烯基碳的SN2(双分子亲核取代)反应;以前人们认为这个反应不能进行,而烯基碘化物7使这个反应成为了可能。碘苯还原消除反应后,烯基碘化物7中的α-氢原子具有足够的酸性可用于α-消除反应的进行,且在一定基质存在的条件下,生成较好产率的亚烷基碳烯。
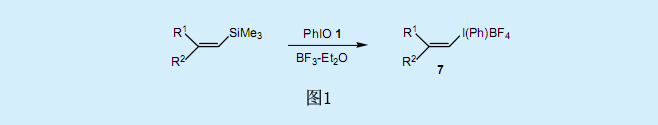
与烯基碘化物的合成方法类似,可以利用炔基硅烷和炔基锡烷合成炔基碘化物8(图2)。炔基碘化物可直接从末端炔基开始进行合成5)。炔基碘化物8和烯醇基阴离子发生迈克尔加成反应形成环戊烯骨架。如图2所示,炔基碘化物8(R=H)是一种良好的乙炔化试剂6)(可以向东京Kasei公司购买)。亚烷基碳烯是反应的中间产物,而炔化产物则可以通过将α-氢中的1,2号氢原子重排获得。
2.丙二烯基碘化物的合成及其还原性碘-克莱森重排反应
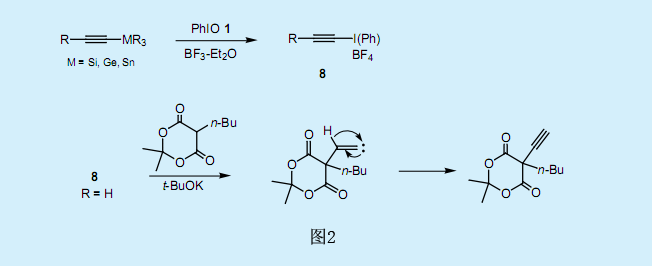
有机硅酮-碘和炔丙基硅烷交换反应的应用催生了由SE2(双分子亲电取代反应)反应生成λ3—炔基碘化物。当碘苯二乙酸2与炔丙基硅烷9在BF3存在条件下反应时,炔丙基被接枝到碘苯分子的邻位上,并生成了高产率的邻炔丙基碘代苯107)(图3)。在研究了若干其他反应之后,我们获得了以下的观察结果:
1)三价碘总是被还原为单加碘。
2)炔丙基化反应总是在碘基团的邻位上发生。
3)新的C-C键会特定性地在丙炔基的α-碳上形成。
4)反应一个较低的温度下进行。
5)丙炔基锗烷和乙炔基锡烷也可用于反应。
6)羟基丙炔基硅烷和乙酸基丙炔基硅烷可用于反应。
综合以上的观察结果,反应机理如图3所示。炔丙基硅烷可与亲电子试剂,例如卤族元素和酰氯物质,发生SE2反应。首先,反应生成了λ3—丙二烯基碘化物11。随后,经分子内的[3,3]单键转移重排反应,炔丙基基团转移至邻位上,接着乙酸基随着炔丙基碘代苯10的形成及12的还原消除反应而被释放出来。λ3—丙二烯基碘化物11进行分子内重排时,其分子性质取决于单价碘烯烃化合物的交叉反应。尽管普通的克莱森重排反应需要加热到150-250℃,但还原性碘-克莱森重排反应在低温下就可以进行。假设反应的速率控控制步骤为λ3—丙二烯基碘化物11的[3,3]单键转移重排反应。由于破坏顶点的碳-碘(Ⅲ)键只需要很小的能量,所以11的碘—克莱森重排反应所需的活化能较低。一般来说,芳香基-碘化物ArIX2具有T形几何形状,超价I(Ⅲ)—X键与芳香基的π键很好的相连。这种有利的轨道相互作用能够促进11的重排反应发生。
在上述的还原性碘—克莱森重排反应中,由于没有λ3—丙二烯基碘化物11生成的直接证据,我们可以认为它是一个中间产物。因此,为了分离和检测中间产物—丙二烯基碘苯14,我们做了二甲基丙炔基硅烷13与亚碘酰基苯甲酸5的反应(图4)。然而看似合理的假设—丙二烯基碘苯的[3,3]单键转移反应却较难进行,这可能由于分子末端的两个甲基的位阻及其电子效应所导致的[3,3]单键转移反应中超价碳—碘(Ⅲ)键断裂却没有和芳香基的π轨道重叠造成的。和预期一样,我们没有在[3,3]单键转移反应中发现邻位的炔丙基化反应。遗憾的是,我们没有检测到丙二烯基碘苯14的生成。与我们的预期相反,该反应生成了过氧烷基碘化物15。过氧碘化物15在同一个分子中含有过氧烷基基团和超价碘(Ⅲ),其均可作为氧化剂,但是在固态时较稳定。X射线衍射分析显示碘原子具有典型的T形几何结构,且具有超价化合物形状上局部变形的特点。
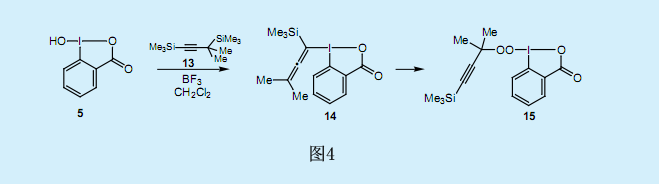
3.过氧化叔丁基碘化物的合成
超价λ3-有机碘化物具有一个作为配位体的过氧烷基基团,而这一结构是非常不稳定的。1968年Milas和Plesnecar报道称,在-80℃的二氯甲烷中过氧叔丁酸可与亚碘酰苯1反应生成过氧化叔丁基自由基和碘苯。假设在该反应中,碘原子上的初始配体交换生成了不稳定的双过氧烷基碘化物,从而导致O-I超价键的均匀断裂,并生成了过氧化叔丁基自由基。由于过氧烷基碘化物15在室温下非常稳定,因此其分离就十分地引人关注了。这可归因于五元杂环化合物的形成所导致的同一位置上顶端杂环配体和中线芳香配体的固定作用。由于易断裂的I-O键与苯基的π轨道间没有的轨道的相互作用,因此这种排列所形成的亚酰碘苯甲基酮就使得过氧烷基碘化物15的稳定性增强。
这种具有独特结构的过氧烷基碘化物预计可用来作为有机合成中的一种新型氧化剂。据此,设计引入过氧化叔丁基并尝试合成碘化物6,以其作为典型化合物。亚酰碘基苯甲酸5在室温下不与过氧叔丁酸发生反应,且由于其活性较差很容易被还原。然而,当反应混合物中加入路易斯酸后,碘原子上的配体交换反应按期望的进行,生成高产率过氧碘化物6(见图5)。邻亚酰碘基苯甲酸5的氧原子上的BF5配位作用使得反应活化。产物过氧化叔丁基碘化物6在固体状态下是很稳定的,且在室温下以晶体状态保存一年以上也未见其分解。

尽管过氧碘化物6在固体状态下是稳定的,但其在溶液中却很容易分解。当过氧碘化物6在室温下溶于氯仿中后,通过配位体交换反应就生成氯碘氧化物,其中过氧碘化物6的半衰期大约为4天。将结晶化的过氧碘化物在140℃下加热,其迅速地分解为1,2-间二碘苯(46%)、碘苯(6%)、邻碘苯甲酸(14%)和丙酮(43%)。在这一吸热分解反应中,由于过氧基团与碘原子间弱超价键的断裂,生成了过氧化叔丁基和9-碘-2σ-碘酰氧基16,进而导致了分解反应的发生。
4.经由过氧化叔丁基碘化物氧化的苄基氧化反应
过氧化叔丁基碘化物6可有效的应用于苄基醚17上的苄基—亚甲基基团的氧化,生成苯甲酸酯类18。这些反应可在室温下的氮气环境中发生。酯类的产量与溶液的介电常数息息相关,尽管室温下这一反应非常慢,但在具有较小介电常数的苯中还是得到了最佳的反应结果。不过,在苯中添加碱金属碳酸盐可以相当程度地加快这一反应。
在有机合成反应中,苄基经常被用来作为醇类的保护基团。由于酯类容易水解成醇类,过氧碘化物6就为苄基提供了一种氧化去保护方式。与去保护反应相关的一类常见问题是它们的化学选择性。苄基位上的选择性氧化甚至在MOM基团、甲硅烷基团、乙酰基团或者四氢吡喃基团存在条件下也会发生。烯丙基也可用来作为醇类的保护基团,而过氧碘化物6则对将烯丙醚类氧化为相应的αβ-不饱和酯类有益。另外,其它芳香烃类也很容易发生苄基氧化,像茚满、四氢化萘、二氯化蒽和茐等都很容易被氧化。这些反应的部分结果如表1所示。
自由基抑制剂像α-生育酚和galvinoxyl可抑制苄基上亚甲基的氧化,这也意味着自由基种类的混乱。为了证实那些自由基产自于苄基位上,用与碳自由基反应迅速的2,2,6,6-四甲基哌啶-N-氧化物(TEMPO)来捕获苄基自由基。我们研究了丁基苄醚17a的氧化反应的取代效果(图6)。对位或间位上吸电子基氯的引入减缓了反应速率,而对位上甲基烷氧基或甲基的引入则加快了反应速率。Hammet相关系数是在相对反应速率和取代常数σ+间建立的,其中ρ=-0.30。这一ρ值可与由苯甲酸基自由生成的二苄基醚中苄基上氢的分离常数ρ=-0..65相比较。氘的主要同位素效应检测结果显示为一个很大的值(KH/KD=12-14)。这一同位素效应也强有力地表明苄基中C-H键的断裂为速率控制步骤。
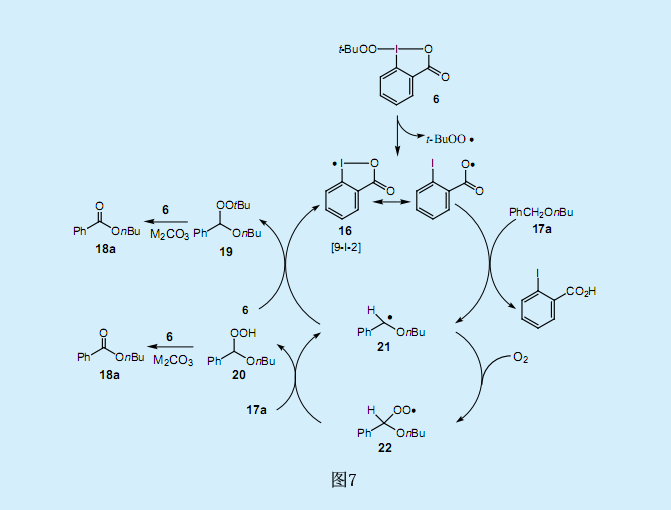
我们也对分子氧对反应的影响作了研究。十分引人关注的是,在过量丁基苄基醚17a(相对于过氧碘化物6而言)存在条件下,氮气氛围下的反应时间延长了达到了410h,并产生了比理论值更多的苯甲酸酯18a(约600%);而在无氧条件下,则生成了24%的苯甲酸酯18a和72%的过氧乙缩醛19(苄基位上含有过氧化叔丁基)。这一结果表明,反应的两种中间产物过氧乙缩醛19和过氧乙羧酸20是由与分子氧的反应产生的。假设两种中间产物均转化为苯甲酸酯,反应机理如图7所示。
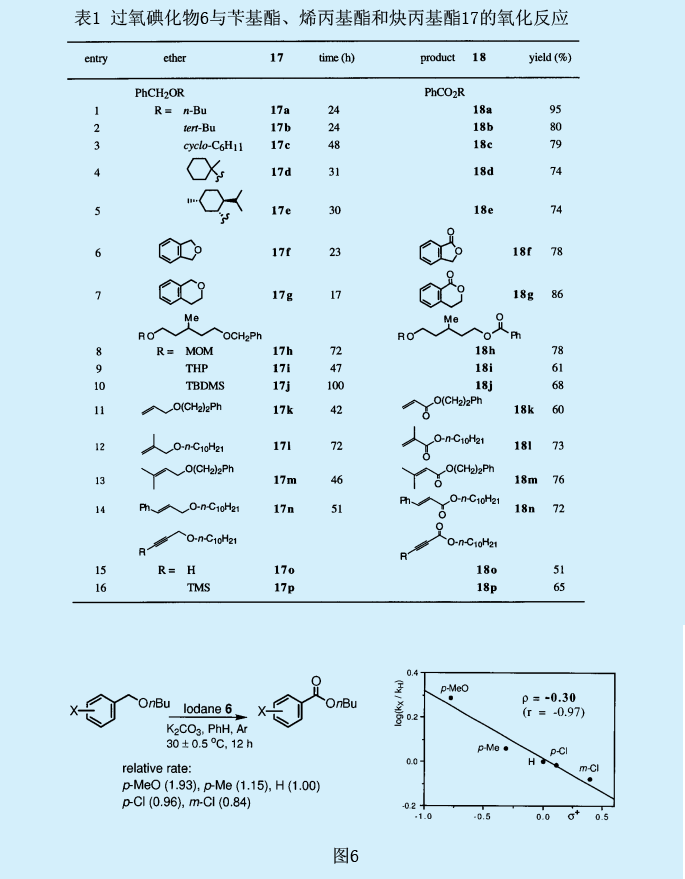
首先,过氧基团的氧原子和过氧碘化物6中碘原子间的弱超价键断裂,生成了过氧化叔丁基自由基和9-碘-2σ-碘酰氧基自由基16。当亲电子的碘酰氧基自由基16吸取苄基醚17a的苄基上的氢就生成了苄基自由基21。苄基自由基21与过氧碘化物6进一步反应生成过氧化叔丁基乙缩醛19,并分解成相应的酯18a。另一方面,当反应系统中存在分子氧时,苄基自由基21与氧反应生成过氧基自由基22。过氧基自由基22与17a的苄基上的氢进一步反应生成过氧乙缩酸20和苄基自由基21。过氧乙缩酸20在一定反应条件下转化为相应的酯18a。
5.经由过氧化叔丁基碘化物氧化的硫化物的氧化反应
过氧化叔丁基碘化物6将硫化物氧化为亚砜。乙腈溶液中二烃基硫化物和烷基芳香基硫化物可以高产率的转化为亚砜(方法A)。二氯甲烷溶液中二烃基硫化物也可以高产率的转化为亚砜(方法C)。在乙腈溶液中添加BF3-Et2O可以加快反应的进行(方法B)。
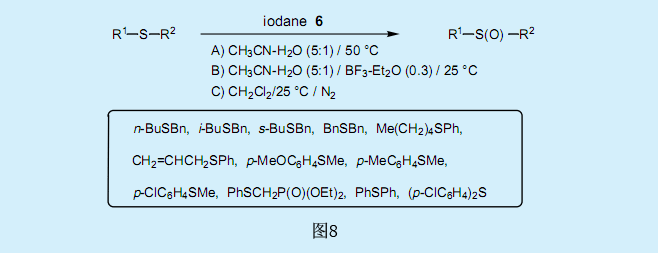
对乙腈溶液中苯甲硫醚取代反应的取代基效应研究表明,ρ对取代常数σ有一个很大的负值(-3.35)。而在乙腈溶液中添加BF3-Et2O后ρ值为-2.23,和取代常数σ+有更好的相关系数。从以下的观察结果显示,乙腈溶液中的反应为离子反应这一假定看来是合理的。
1)过氧碘化物6与亚酰碘苯甲酸5可达到共存平衡。
2)单一的以亚酰碘苯甲酸5或过氧化叔丁酸自身为氧化剂反应不能发生,但是当两者共同使用时反应可以发生。
3)另外,添加自由基去除剂galvinoxyl对反应影响很小。
此外,这也充分地表明过氧化叔丁基碘化物6是活性物种,且在硫原子上产生了大量的带有正电的活性中间体。另一方面,二氯甲烷(方法C)中添加galvinoxyl后完全抑制反应的发生表明,该反应的机理为自由基机理。
二硫缩醛的去保护氧化反应也会发生。将二硫缩醛与过氧碘化物6在乙腈中进行处理,发现它们能在数分钟内完全反应并生成高产率的酮。过氧碘化物6可用来作为将硒醚氧化为硒亚砜和膦类氧化为膦氧化物的氧化剂。此外,过氧碘化物6也可以将2mol的三苯基膦氧化为相应的氧化物。
6.经由过氧化叔丁基碘化物氧化的胺类的氧化反应
过氧化叔丁基碘化物6可以有效的氧化胺类有机物。仲胺与过氧化叔丁基碘化物6发生脱水反应生成亚胺类有机物。碳酸钾的添加可以加快反应的进行。四氢异喹啉氧化后可获得高产率的二氢异喹啉。当使用过量的过氧碘化物6时则生成异喹啉。当过氧碘化物6与季胺类有机物反应时则生成过氧氨基缩醛,其中过氧基取代添加在胺的α碳原子上。

7.经由过氧化叔丁基碘化物氧化的酚类的自由基氧化反应
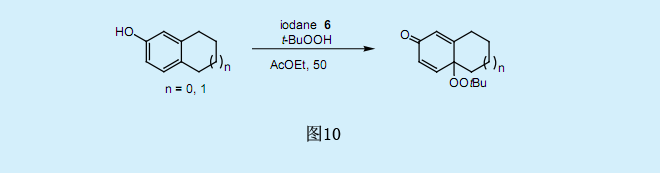
过氧化叔丁基碘化物与对位烷基取代的酚反应生成4-(过氧化叔丁基)环己二烯酮。在过氧化叔丁酸中,过氧碘化物6与对位取代的酚在温和条件(乙酸乙酯/50。C)下反应生成高产率的过氧化叔丁基环己二烯酮。由于在galvinoxyl存在下氧化反应基本上被完全抑制了,而仅有少量副产物过氧化叔丁基环己二烯酮二聚物生成,因此我们假设这一反应为自由基反应。经由共振稳定的苯氧基自由基是一种反应中间体,它与过氧化叔丁基自由基耦合生成过氧化叔丁基环己二烯酮。
8.结论
在过氧化叔丁基碘化物6中,过氧化叔丁基与三价碘原子由超价键连接。大量关于其结构的调查研究使我们对其具有很高的活性很是期待,但其潜在的爆炸风险也令人担忧。很可能仅有少数的化学家尝试合成这种化合物。相反,令人惊讶的是结晶态的过氧化叔丁基碘化物6是十分稳定的,且在室温下不会分解。另外,除非将其配制成溶液(但溶液中的反应也十分缓慢),否则组成超价键的自由基不会断裂。一般来说,该反应在50℃以下才会进行,而超过这一温度下的反应至今无人尝试。
如上讨论所言,过氧化叔丁基碘化物的开发是由我们集中力量寻求一种新的碘的潜在利用方式得到的。这些结果是由Takao Ito博士(日本烟草研究实验室)精心实验得到的。笔者在此衷心地感谢为此做出贡献的学生们,他们的名字在本文所列的参考文献中指出。
参考文献:
1) a)G, F. Koser, in "The Chemistry of Functional Groups, Supplement D2"; Ed. by S. Patai and Z. Rappoport, Wiley, New York (1995); Chapters 21. b) A. Varvoglis, "The Organic Chemistry of Polycoordinated Iodine", VCH, New York (1992).
2) M. Ochiai, T. Ito, Y. Masaki, M. Shiro, J. Am. Chem. Soc., 114, 6269 (1992).
3) M. Ochiai, Kagaku Sosetsu, "Hypervalent Organic Compounds", 34, 181 (1998).
4) M. Ochiai, M. Toyonari, T. Nagaoka, D.-W. Chen, M. Kida, Tetrahedron Lett., 38, 6709 (1997).
5) M. Ochiai, in "Chemistry in Hypervalent Compounds"; Ed. by K. Akiba, Wiley-VCH, New York (1999); Chapters 12.
6) M. Ochiai, T. Ito, Y. Takaoka, Y. Masaki, M. Kunishima, S. Tani, Y. Nagao, J. Chem. Soc., Chem. Commun., 1990, 118.
7) M. Ochiai, T. Ito, Y. Takaoka, Y. Masaki, J. Am. Chem. Soc., 113, 1319 (1991).
8) M. Ochiai, T. Ito, M. Shiro, J. Chem. Soc., Chem. Commun., 1993, 218.
9) M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto, M. Shiro, J. Am. Chem. Soc., 118, 716 (1996).
10) M. Ochiai, A. Nakanishi, T. Ito, J. Org. Chem., 62, 4253 (1997).
11) M. Ochiai, D. Kajishima, T. Sueda, Heterocycles, 46, 71 (1997).
12) M. Ochiai, A. Nakanishi, A. Yamada, Tetrahedron Lett., 38, 3927 (1997).
注:本文为提供者翻译的,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号