脂质体毛细管电泳研究进展
- 2012-05-15
- 专题
自1965年由英国的Bangham等(1)首先发现磷脂在水中可以自发形成脂质体(Liposome)以来,对脂质体的实验研究日渐广泛,已遍及生命科学、膜工程学等领域,并逐渐向临床应用发展。脂质体是由磷脂和其它两亲性物质分散于水中形成的由一层或多层同心脂质双分子膜包封而成的球状体,具有亲水性和疏水性两性性质。依其所含脂质双分子层的层数,脂质体可分为单室脂质体和多室脂质体。其中单室脂质体又可根据粒径的大小分为小单室脂质体(粒径范围0.02 ~0.08 μm)和大单室脂质体(粒径范围0.1~1μm)。
随着生物膜色谱(Biomembrane Chromatography)的发展,脂质体作为一种分析工具受到了越来越多的关注,并成为近年来较为活跃的研究领域(2,3)。生物膜色谱是一种新型液相色谱模式,它以天然或人工模拟生物膜为固定相,主要用来研究溶质分子与生物膜间的相互作用。目前,最常用的生物膜色谱固定相是固定化脂质体(4-6),因为它能更好地模拟生物膜的脂双层结构,并具备生物膜的流动性特征。这种人工模拟的生物膜体系不仅可以精确地模拟生物膜的化学环境,而目其物理性能也可以通过调节温度、pH、离子强度等得到有效控制。
固定化脂质体色谱具有许多优点,但是它的应用并不广泛,主要原因是制柱比较困难、成本高。其次高效液相色谱(High Performance Liquid Chromatography ,HPLC)常用的硅胶基质及其化学键合固定相的pH适用范围较窄(2~8),色谱柱易被污染且难再生,流动相需要大量有机溶剂,成本较高,且污染环境。与固定化脂质体色谱相比,脂质体毛细管电泳(Liposome Capillary Electrophoresis ,LCE)更有优势,其具有制备简便、溶剂消耗少、进样量小、成本低等优点。1995年Zhang等(7)首次提出了LCE的概念,近年来,LCE已经得到了较快发展,尤论是在方法学方面,还是在应用方面都有了比较深入的研究。
1脂质体的制备
目前,脂质体的制备技术己经相当成熟,有关报道和综述也较多(8-10)。常用脂质体制备方法有薄膜分散法、注入法、超声分散法、冷冻干燥法、冻融法、逆相蒸发法、复乳法等。
LCE中所用的脂质体主要是单室脂质体。多室脂质体由于背景噪声大,灵敏度低而不适合。与固定化脂质体色谱相比,LCE中的脂质体制备方法比较单一,通常采用薄膜-超声分散法和薄膜-挤压分散法。薄膜-超声分散法是制备单室脂质体的最早方法,即将磷脂、胆固醇等类脂质溶于有机溶剂中,旋转蒸发除去有机溶剂,使在烧瓶内壁形成薄膜,然后将水溶性介质加入烧瓶中并不断振荡,得到多室脂质体,最后经超声处理得到单室脂质体,其粒径大小依超声时间的长短而不同。薄膜-超声分散法的缺点是其制备的脂质体粒径分布不太均匀,内层的磷脂分子数要小于外层的磷脂分子数,热力学不稳定,脂质体之间容易发生融合(11)。薄膜-挤压分散法是目前制备单室脂质体的最常用方法。这种方法也是先采用薄膜法制得多室脂质体,然后在一定压力(<7.091×l05Pa)下使大多室脂质体通过很小孔径的聚碳酸醋膜,以获得大小均匀的单室脂质体(12)。
2 LCE的分类及脂质体涂层的制备
LCE大体可分为两类。一类从电泳缓冲液入手,将脂质体作为添加剂,直接加入电泳缓冲液中,形成假固定相,其基本原理与胶束电动毛细管色谱(Micellar Electroliinetic Capillary Chromatography,MECC)类似,因而被称之为“脂质体电动毛细管色谱(Liposome Electrokinetic CapillaryChromatography LECC)" (7,13~21)。另一类从毛细管管壁入手,将脂质体作为涂层剂,采用氢键吸附法(22~27)、静电吸附法(28,29)、抗生物素蛋白-生物素亲和法(30)等固定化方法,使毛细竹内壁形成较稳定的脂质体涂层,因而可称之为“固定化脂质体毛细竹电泳(Immobilized Liposome Capillary Electrophoresis, ILCE) "。两者相比,ILCE的柱效较低,但其脂质体的消耗少,并可与质谱联用(2)。
ILCE的脂质体涂层是磷脂双层(涂层在毛细管内壁的脂质体破裂,相互融合)还是囊泡层(涂层在毛细管内壁的脂质体形态完整),取决于多种因素如脂质体的制备方法、粒径大小、载体材料及浓度、电泳缓冲液中Ca2+存在与否等(11,31,32)。脂质体涂层的稳定性与固定化方法有很大关系。氢键吸附法、静电吸附法等非共价键合的方法,操作过程简单,但脂质体涂层稳定性差,流失严重;亲和法则使脂质体涂层的稳定性得到明显改善,缺点是制作成本相对较高。下面对这些固定化方法进行一一介绍。
2.1氢键吸附法
该法十分简便,只需将含有脂质体的电泳缓冲液与毛细管平衡数分钟,此时由于脂质体中磷脂的一P=O基团与融熔石英毛细管内壁的硅烃基产生氢键作用力,而使脂质体充分吸附于毛细竹内壁,然后用不含脂质体的电泳缓冲液冲洗,除去未吸附的脂质体即可。但是对于负性脂质体而言,该法不太稳定,因为毛细管内壁的硅烃基会与负性脂质体发生相互排斥作用。
Hautala等(22)选用平均粒径大约100nm的大单室负性脂质体(组成成分:1-棕相酞-2-油酸-3-磷脂酞胆碱/磷脂酞妊氨酸/胆固醇,POPC/PS/Cholesterol),通过氢键吸附法制备了LCE的脂质体涂层。具体涂层过程为:0.5mol/L盐酸冲洗裸露熔融石英毛细管10min→水冲洗15min→电泳缓冲液冲洗5min→含有脂质体的电泳缓冲液冲洗10min→含有脂质体的电泳缓冲液在毛细竹内预稳定15min→电泳缓冲液冲洗除去未吸附的脂质体。在整个涂层过程中,电泳缓冲液的种类保持相同。Hautala等发现,脂质体涂层的最终效果和稳定性受到冲洗脂质体溶液的时间、分析电压、电泳缓冲液的种类等因素的影响,其中电泳缓冲液的种类对负性脂质体涂层的涂层效果影响最大;采用三(三烃甲基氨基甲烷)缓冲液、磷酸欲缓冲液和麦黄酮(N-三烃甲基甲基甘氨酸)缓冲液制备的脂质体涂层,电渗流((EOF)有少许增加(1%~7%),而用HEPES [N-(2-羟乙基)哌嗪-N-(2-乙磺酸)]缓冲液制备的脂质体涂层,EOF减少24%,这表明在HEPES缓冲液中,脂质体与毛细竹内壁的作用最强,因而得到最好的涂层效果。Wiedmer等(27)尝试用一系列与HEPES结构相似的化合物(均含呱嗦基团)溶液作为电泳缓冲液,结果发现采用这些缓冲液制备的负性脂质体(组成成分:磷脂酞胆碱((PC)/PS)涂层也相当稳定。另外,在电泳缓冲液中加入一价金属离了(如Ca2+,Mg2+,Zn2+)或电泳温度在相变温度以下时均能增加磷脂膜的刚性,提高脂质体涂层的稳定性(24,25)。
2.2静电吸附法
先将带电荷的化合物涂布于毛细竹内壁,然后将带相反电荷的脂质体填充于毛细竹中,则脂质体依靠静电作用力吸附于毛细竹内壁。因为静电作用力比氢键作用力大,所以这种方法制备的脂质体涂层比氢键吸附法制备的脂质体涂层更稳定一些。
Ornskov等(30)先将琼脂糖衍生物(含有带正电荷的铵离了)涂布于毛细竹内壁,然后用含有负性脂质体(组成成分:POPC/PS)的溶液冲洗毛细管5min,则负性脂质体通过静电作用力吸附在带正电荷的毛细管内壁。实验发现,随着涂布于毛细管内壁的琼脂糖衍生物电荷密度的增加,固定在毛细竹内壁的脂质体含量增大,一系列非离子化合物在ILCE中的保留值增大。这证明在脂质体的涂层过程中,静电作用力确实起主要作用。
2. 3抗生物素蛋白一生物素亲和法
首先将抗生物素蛋白共价键合到氨基化的毛细竹内壁,然后将生物素酞基化的脂质体填充到键合有抗生物素蛋白的毛细管内(如图1)。通过抗生物素蛋白-生物素的亲和作用力将脂质体固定在毛细管内表面。由于溶解脂质体的电泳缓冲液中含有抗生物素蛋白,因而单个生物素酞基化的脂质体之间也可通过亲和作用力发生聚合。与氢键吸附法制备的脂质体涂层相比,亲和法制备的脂质体涂层其稳定性有了相当程度的提高,因为抗生物素蛋白与生物素间的亲和作用力比氢键作用力强得多。在涂层过程中还发现,毛细管内壁覆盖了4~15层脂质体,且采用大单室脂质体制备的脂质体涂层所固定的磷脂含量要比采用小单室脂质体制备的脂质体涂层所固定的磷脂含量高出2倍。

3 LCE的应用
LCE的应用的应用领域主要分为二个方面:(1)在各种有机化合物及多肤、蛋白质分离中的应用;(2)在对映异构体分离中的应用;(3)在药物一生物膜相4.作用研究中的应用。
3.1 LCE在各种有机化合物及多肤、蛋白质分离中的应用
LCE中,溶质的分离主要基于不同溶质在电泳缓冲液和脂质体 (LECC中为假固定相,ILCE中为涂层)之间的分配系数不同而进行的。溶质的带电量、化学结构、极性、甚至浓度都会影响溶质的分配系数大小(33,34)。另外,在分配过程中,不同溶质在脂质体中的分配部位也可能不同(极性区域或疏水区域) (35)。
目前,LCE已用于苯的衍生物及苯酚类化合物(14,15,18,25)、类固醇(15~17,22,25)、多肤和蛋白质(7,19~21,23,24,28)、β-阻断剂(18)、其它药物(7,13,18,26,27)等的分离。Wiedmer等(16,17)在电泳缓冲液中添加负性脂质体分离了8种中性皮质类固醇化合物(17-异醛固酮、醛固酮、21-脱氧皮质醇、皮质酮、雄烯二酮、睾酮、黄体酮和17-烃孕酮)。脂质体中总脂质的含量、脂质体组成中不同磷脂的摩尔比、脂质头部基团的种类、电泳缓冲液的种类以及脂质体的物理状态(胶态和液品态)等多种因素都会影响分离效果(16)。Wiedmer等(17)分离衡体激素时还发现,脂质体组成成分中胆固醇的含量对类固醇化合物的分离有很大影响;胆固醇含量越高,化合物的分离度越大。
CE分离分析多肤和蛋白质时,样品的吸附严重影响了分离分析结果的重现性。这种吸附主要是带部分正电荷的多肤和蛋白质分子与带负电荷的熔融石英毛细竹内壁相互作用的结果。而LCE分离分析多肤和蛋白质时,由于毛细竹内壁吸附了脂质体,硅烃基被掩蔽,样品的吸附减少,因而使柱效增大,电泳峰形和分离分析结果的重现性明显改善;同时,多肽、蛋白质与脂质体之间的相互作用使分离选择性得到提高(19,20,23)。 Corradini等(19,20)在电泳缓冲液中添加POPC脂质体分别分离了4种碱性蛋白(细胞色素C,溶菌酶,核糖核酸酶A, a-胰凝乳蛋白酶原)和3种合成多肤(血管紧缩素I,Ⅱ,Ⅲ);研究发现,毛细竹内注入的脂质体溶液的体积或脂质体载体材料(磷脂)的浓度增加,多肤的迁移时间都增加,分离度提高(20)。Cunliffe等(23)采用ILCE(脂质体组成:二月桂酞磷脂酞胆碱/Ca2+,DLPC/Ca2+),在pH3~10范围内成功分离了阴性和阳性蛋白质,理论塔板数高达1400000/m迁移时间的日内精密度小于1.3%,日间精密度小于4%,回收率达到93%以上。
3.2 LCE在对映异构体分离中的应用
在对映异构体的CE分离中,一般采用在电泳缓冲液中加入乎性试剂的方法。很多天然生物大分子如牛血清蛋白、人血清蛋白、糖蛋白、溶菌酶等本身含有不对称碳原子,能够对许多小分子化合物产生乎性识别作用,是一类高效、廉价、易得的乎性试剂(36)。但是这类天然生物大分子在紫外光区都有强烈的吸收,因而大大限制了其应用范围。将这类乎性试剂固定在毛细竹内壁是解决此问题的主要方法。Bo等(28)首次将溶菌酶固定在毛细竹内壁脂质体涂层的磷脂膜上,在电泳缓冲液中不含乎性试剂的情况下,成功分离了D-和L-色氨酸。他们在涂层过程中发现,若将负性脂质体直接涂敷在裸露的毛细竹内壁,则涂层的稳定性很差。为解决该问题,Bo等先在毛细竹内壁涂敷M1C4(1-(4-碘丁基)-1,4-甲基哌嗪-1-碘化铵),然后再涂敷负性脂质体,最后用溶菌酶溶液冲洗涂层后的毛细竹。因为 M1C4掩蔽了带负电荷的硅经基,减少了负性脂质体与硅经基的排斥作用,而目与负性脂质体静电相吸,所以脂质体涂层的稳定性大大提高。溶菌酶由于较易渗入到磷脂膜内,而被比较稳定地固定于毛细竹内壁。与此同时,Bo等还尝试将M1C4涂敷于毛细竹内壁后直接涂敷溶菌酶,虽然D-和L-色氨酸的分离度提高了,但却以牺牲稳定性为代价,分离结果的重现性很差。
3.3 LCE在药物与生物膜相互作用研究中的应用
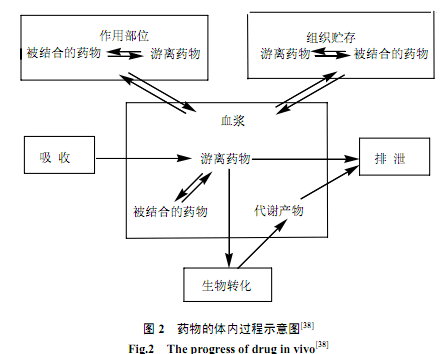
药物在体内的吸收、分布和排泄统称为药物转运,而在体内发生的化学变化,则称为药物代谢或生物转化。在这些过程中,各种因素的相互影响如图2所示(37)。因为人体内的各部分之间彼此有生物膜隔开,所以生物膜是影响药物体内过程的最主要因素之一。药物的活性、毒性、体内分布及其它生理过程都与药物在生物膜上的分配状况相关(38),因此测定药物在生物膜上的分配系数对药物设计和药物筛选来说十分重要。由于LCE提供了一个人工模拟的生物膜表面,能够模拟药物结合到生物膜表面或分配到生物膜脂双层时发生的疏水、静电等相互作用,因而药物在LCE中的迁移行为和选择性能够直接反映药物在生物膜上的分配状况。
药物在LCE中的迁移行为取决于EOF、药物与脂双层的相互作用、磷脂(或其它脂类尤其是胆固醇与磷脂的比例)的用量等诸多因素。为了表示药物与脂双层的相互作用,可参照色谱原理,假定一个与EOF和磷脂的用量无关的容量因子(见式((1))(7)。
k'=(t- t') /( t'×B) (1)
式中k'为容量因子;t为药物在脂质体毛细竹电泳中的迁移时间;t'为药物在无脂质体毛细竹电泳中的迁移时间;B为磷脂的用量。
通过式(2)还可以将药物在LCE中的容量因子k'与药物的脂质体一水分配系数K1w联系起来。
k'=φK1w (2)
式中φ为相比,等于LCE中脂质体与电泳缓冲液所占的体积比。
一个药物在生物膜上的分配情况通常用该药物在正辛醇和水中浓度比值的对数(lgPow)来衡量(19,39)。然而正辛醇与生物膜有很大差异,用lgPow评价药物与生物膜的相互作用是不够准确的。脂质体的结构与生物膜很相似,因此,K1w能更准确地反映药物与生物膜的相互作用。
由式(2)可知,K1w与相比φ有关。在LCE中,φ是一个反映脂质体本质特征的参数,当脂质体的组成恒定时,φ不变,且能较准确地计算出来。在HPLC中,φ是固定相与流动相所占的体积比,因而不同的色谱柱,φ的差异性很大,且不易计算(18)。因此,与固定化脂质体色谱相比,采用LCE测定K1w更准确,重现性更好。
Burns等(14,18)首次将LCE技术应用于定量测定药物的K1w。研究表明,LCE测定的K1w与lgPow、线性溶剂化能量关系式(Linear Solvation Energy Relationship, LSER}是定量结构分配关系的一种表现形式)所预测的化合物的K1w值都有很好的相关性。这说明脂质体的溶剂化特性与正辛醇相似,并且K1w还可应用于定量结构分配关系(Quantitative Structure-Partition Relationship, QSPR)方面的研究。
药物的吉布斯自由能变化△Go即药物在无脂质体毛细竹内的吉布斯自由能与在脂质体毛细竹内的吉布斯自由能之差,是评价药物与生物膜间相互作用的另一个重要参数(7,13,26)。△Go越小,说明药物与生物膜间的相互作用越强,药物更易进入生物膜内。但△Go的计算公式比较复杂,为此,通常选择一种参比药物(如阿司匹林),并假设EOF为零,通过计算0(OCe>见式(3)来衡量药物的△Go(7)。
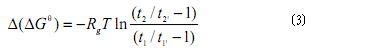
式中Rg为标准气体常数;T为热力学温度;t1为参比药物在脂质体毛细管电泳中的迁移时间;t1’为参比药物在无脂质体毛细管电泳中的迁移时间;t2为被测药物在脂质体毛细竹电泳中的迁移时间;t2’为被测药物在无脂质体毛细竹电泳中的迁移时间。
Manetto等(13,26)分别采用LECC和ILCE(脂质体组成成分:POPC)测定了一系列不同极性药物的△(△Go),对这些药物与脂质体间的相互作用进行了探讨,并比较了两种方法测定结果的差异性。在LECC中,他们选用聚酞亚胺涂层毛细竹(总长30cm,有效长度19.5cm,内径50μm),样品从负极端进样;电泳缓冲液pH9.2时,水杨酸、阿司匹林、酮基布洛芬、苯妥英和普蔡洛尔5种药物全部与脂质体发生相互作用,但测得的水杨酸的△(△Go)值比文献[7](采用LECC)测得的水杨酸的△(△Go)值低0.2 kJ/mol(约17%) ,可能因为电泳缓冲液pH9.2时仍有残留的EOF而导致测定结果偏低,因为扣除EOF后计算得到的△(△Go)与文献[7]的测得值很接近(13)。在ILCE中,选用未涂层的毛细竹,测得的水杨酸的从△(△Go)值(扣除了EOF的影响)比LECC测得的值高出5%(26)。
一般来说,在利用LCE研究药物与生物膜的相互作用时,为了使脂质体与人体生物膜具有更大的相似性,电泳缓冲液pH的选择应依据相应的人体生理pH(如小肠中生理pH约5.4,细胞外生理pH约7.0,细胞内生理pH7.4)而定。另外,由于pH会影响脂质体载体材料的水解速度(pH过大或过小,磷脂的水解速度都将增大(40),因此LCE应使用新鲜制备的脂质体。
4展望
LCE作为一种新颖的分离分析方法,目前仅有相关的研究报道。LCE除在分离分析方面具有一定潜力外,更重要的是脂质体与生物膜结构类似,利用LCE研究药物与脂质体间的相互作用,能最大程度地反映药物分子与生物膜相互作用的真实情况。这种体外实验模拟体内条件的筛选模型为药物筛选和药物设计提供了强有力的工具。再者,LCE与生物膜色谱相比,方法简单,可操作性强,成本低廉,尤其是测定药物的K1w准确、重现性好,具有更好的发展前景。目前,LCE无论在理论还是技术方面都还不太成熟,应用范围也较为有限,因而其发展空间还很广阔。
参考文献
(1)A D Bangham, MM Standisli, J C Watkins. J. Mol. Biol., 1965, 13:238252
(2)A C6mez-Hens, J M Femandez-Romero. Trends in Ana1. Chem,2005,24(1):919
(3)S K Wiedmer, M-L Riekkola, M S Jussila. Trends in Anal. Chem., 2004, 23(8):562582
(4)Q Yang, X-Y Liu, M Yoshimoto et al. Anal. Biocliem,1999, 268:354362
(5)Q Yang, X-Y Liu, S-I Ajiki et al. J. Chromatogr. B, 1998, 707:131141
(6)E Kranse, M Dathe, T Wieprecht et al. J. Chromatogr. A, 1999, 849:125133
(7)Y X Zhang, R Zhang, S Hjeren et al. Electrophoresis, 1995, 16:15191523
(8)胡景莲,张辉.中国临床医药研究杂志.2004, 113:1187211874
(9)曹宁宁,刘金鹏.天津理工学院学报,2003, 19(1):3035
(10)赵海霞,郭兴奎,孔德亮等.山东中医杂志,2000, 19(7):435437
(11)I Reviakine, A Brisson. Langmuir, 2000, 16:1806}}1815
(12)高申.现代药物新剂型新技术.北京:人民军医出版社,2002:213
(13)G Manetto, M S Bellini, Z J Deyl. J. Cbromatogr. A, 2003, 990:205214
(14)S T Bums, A A Agbodjan, M G Khaledi. J. Cbromatogr. A, 2002, 973:167176
(15)S K Wiedmer, J Hautala, J M Holopainen et al. Electrophoresis, 2001, 22:13051313
(16)S K Wiedmer, J M Holopainen, P Mustakangas et al. Electrophoresis, 2000, 21:31913198
(17)S K Wiedmer, M S Jussila, J M Holopainen et al. J. Sep. Sci, 2002, 25(7):427437
(18)S T Bums, M G Kbalehi. J. Pbann. Sci, 2002, 91(7):16011612
(19)D Corradini, G Mancini, C Bello. J. Chromatogr. A, 2004, 1051:103110
(20)D Corradini, G Mancini, C Bello. Chromatogr. Supplement, 2004, 60:S 125}S 132
(21)C Bello, G Mancini, D Corradini. Cbimica a 1'Industria (Milan, <st1:country-region w:st="on">Italy), 2004, 86(7):7883
(22)J T Hautala, M V Linden, S K Wiedmer et al. J. Chromatogr. A, 2003, 1004:8190
(23)J M Cunliffe, N E Baryla, C A Lucy. Aual. Chem,2002, 74:776783
(24)M V Linden, S K Wiedmer, R M S Hakala et al. J. Chromatogr. A, 2004, 1051:6168
(25)J T Hautala, S K Wiedmer, M-L Riekkola. Anal. Bioanal. Chem., 2004, 378(7):17691776
(26)G Manetto, M S Bellini, Z Deyl. J Chromatogr. A, 2003, 990:281289
(27)S K Wiedmer, M Jussila, R M S Hakala et al. Electrophoresis, 2005, 26:19201927
(28)T Bo, S K Wiedmer, M-L Riekkola. Electrophoresis, 2004, 25:17841791
(29)E ornskov, S Ullsten, L Soderberg et al. Electrophoresis, 2002, 23(19):33813384
(30)Q Yang, X-YLin, JMiyake. Supramol. Sci,1998, 5(5-6): 769772
(31)R Rapuano, A M Carmona-Ribeiro. J. Colloid Interface Sci., 1997, 193: 104111
(32)A S Muresan, K Y C Lee. J. Pbys. Chem. B, 2001, 105: 852855
(33)M Alumed, J S Burton, J Hadgraft et al. Biochem. Pbarmacol., 1980, 29: 23612365
(34)K Kitamura, H Kano, K Yoneyama. Mol. Phannacol., 1981, 20:124127
(35)A M S Ahmed, F H Farab, I W Kellaway. Pharm. Res., 1985, 2:119124
(36)J Haginaka. J. Cbromatogr. A, 2001, 906:253273
(37)王钦茂.药理学.上海:上海科技出版社,1994:265
(38)C Pidgeon, S Ong, H-L Liu et al. J. Med. Chem., 1995, 38: 590594
(39)郭宗儒.药物化学总论.北京:中国医药利一技出版社,2003:171
(40)M Grit, W J M Underberg, D J A Crommelin. J. Pbarm. Sci., 1993, 82(4): 362366
注:本文为提供者整理翻译,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号