1,1-二甲(基)丙烯-钯复合物:制备与合成潜力
- 2012-05-23
- 专题
前言
由于环丙烷环不一样的键合和固有的环张力(27.5kcal·mol-1),所以在性质和反应上它是碳环中独特的一员。它的化学反应性非常类似于烯族双键;实际上,二个基团都与相邻的电子系统和p电子中心相互作用,并经过加酸、卤素、臭氧,经历催化加氢和环加成,形成金属复合物等……1环丙烯衍生物不仅提供前所未有的合成潜力的构建模块,而且它们被赋予了大范围的生物学特性,包括从抗菌,抗病毒,抗真菌,杀虫,荷尔蒙,神经化学,抗肿瘤活性到酶抑制。2作为机理探针和新药的设计上广泛使用很多生物合成工艺中的关键中间体。2,3因此,对于合成有机化学家和生物有机化学家,包含这些化合物的环丙烯环具有极为普遍的兴趣。
环丙烯基环上的成分可以强烈的影响它的具体反应。因此,邻近给体-受体取代的环丙烯环经历开环反应,得到复合功能化合物或碳环和杂环系统,4然而给电子基团和相邻的多重键在同一碳原子上取代,三元环可以经历立体定向的环扩张,许多有效的合成方案已经包含了这一过程。5,6最近,从钯(0)催化的1-(L-烯基)环丙基酯的亲核取代反应中已经研究出环丙烷环化学反应的新的,有希望的区域,它涉及了π-1,1-二甲(基)丙烯-钯复合物中间形成。
制备
不管是从1,3-二氯丙酮、8环丙酮半缩醛、91-羟基环丙烷羧酸、10还是从有机卤化物和2-烯基羧酸盐的钛(Ⅳ)催化反应中,11,12都是很方便得到1-(1-烯基)环丙醇2a的。然后,通过乙酰氯、氯甲酸乙酯、三氟乙酸酐、酰基或mesyl氯,2a的酯化反应容易生成烯丙基酯2b-f。7,12,13另外,通过1-氯乙烯基碳烯加成到烯烃,14或从1-氯-1-三氯乙烯基环丙烷,15可以制备出1-氯-1-乙烯基环丙烷2g。同分异构乙酸盐3b和碳酸盐3c从2-环亚丙烷基乙醇3a酯化中得到,7或者很容易从环丙酮半缩醛中得到。9,16,17
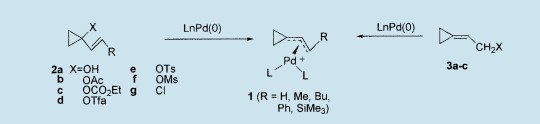
丙烯基乙酸盐和碳酸盐与钯(0)π-丙烯基钯复合物一起生成,它们能够与亲核试剂在原位反应。在特别温和的条件下,这些过渡金属催化碳-碳或碳-杂原子键的形成允许获得大量的丙烯基取代基。然而,在通常用于丙烯基乙酸盐取代反应(通过软亲核试剂(稳定的阴离子)等)的典型环境下,四(三丙基膦)钯(0)[Pd(PPh3)4]存在时,在四氢呋喃中加热回流5天,乙酸盐2b(R=H)不和二乙基2-Na丙二酸酯发4生反应。但是,Pd(PPh3)4存在于四氢呋喃中并回流36小时,同形异构的丙烯基羧酸盐3b与4反应,并以80%的收率生成二乙基(2-环丙基 )丙二酸酯5。7

1(R=H)的形成,至少部分地把一个正电荷给三元环,由于环丙基阳离子(SE=42kcal·mol-1)比环丙基环(SE=27.5kcal·mol-1)更紧,因此2b(R=H)中乙酸盐键的裂解不受化学亲睐。另一方面,来自乙酸盐3b(SE=40.9kcal·mol-1)的1(R=H)的形成只是稍微增加了环张力。对于2b(R=H)与亲核试剂4在四氢呋喃中回流24h的反应,Pd(0)复合物的使用在原位生成了二(二亚苄基丙酮)钯[Pd(dba)2]和1,2-二(二丙基膦)乙烷(dppe),并以31%的收率生成了5,以及此外的一个开环副产品(6%);长反应时间(48h,65℃)导致了低选择性的反应。7在中性环境下,19即使在Pd(dba)2/dppe存在时,碳酸盐2c(R=H)也不与二乙基丙二酸酯反应;与此相反,同分异构的碳酸盐3c以31%的收率得到5(rt,4h)。较好的离去基增加反应活性;因此,三氟乙酸盐2d(R=H)在23[Pd(PPh3)4,65℃,48h]中发生取代反应并得到55%的收率[Pd(dba)2/dppe];而巧妙的是,不管钯(0)配体是什么样的性质,用4更快地取代磺酸盐2e,f(R=H),并在环境温度下5min内以84-86%的收率仅生成二甲基丙二酸酯5。7在室温下24h内,氯化物2g(R=H)也经历Pd(0)催化取代反应,并依靠二 双膦配体,双二丙基膦乙烷或1,4-二(二苯基膦)-甲苯(dppb),分别以51%和66%的收率产生5。20
从1(R=H)的取代反应(通过合适的亲核试剂)中以较高的收率(72-93%)获得(2-环亚丙基乙基)丙二酸酯和相关化合物6-12;而从1(R=Me)和1(R=Rh)分别得到丙二酸酯13(89%)和14(94%)。在[18]-冠(醚)-6、二苄胺、苯甲酮亚胺和苄氧基胺存在时,对于1(R=H)的亲核取代,使用乙酸钾很容易生成(2-环亚丙基乙基)-乙酸盐15(80%)和烯丙基胺衍生物16-18(84-95%)。7,21

亚烷基环丙烷具有非凡的合成潜力,形成了一类特有的紧烯族化合物。因此,它们经历过渡金属催化开环反应,与烯烃和炔底物的[3+2]环加成作用,与硝酮的[1,3]偶极环加成作用,与乙炔二钴羰基络合物的Pauson Khand环合反应。22最近,在广受评议的众多亚烷基环丙烷合成中,23这种新的方法迄今为止表现最好,符合了化学性、地域性、立体选择性和原子经济性的所有要求。24在1-乙烯基环丙基甲苯磺酸盐2e(R=H)和1,1-二甲基丙烯基乙酸盐19之间或环亚烷基乙基乙酸盐3b和3,3-二甲基丙烯基乙酸盐20之间的竞争实验,它们与4的Pd(0)催化反应生成19:1和99:1的5的混合物(>95%)以及同分异构的丙二酸酯21和22(比例7/3)。
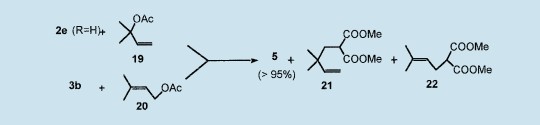
对于复合物1(R=H)和相应的π-1,1-二甲基丙烯基钯复合物(产生于19和20,),已经考虑了不同的结构以解释这样意外的行为。高水平的计算(STO-3G,6-31G和6-31*)已经指出一个较高正电荷在1-乙烯基环丙基阳离子23的初级碳上,然而MINDO/3,AM1和从头计算法预测出一个较高的正电荷在1,1-二甲基丙烯基阳离子24的第三中心上。7计算显示π-丙烯基钯复合物23’和24’有两分子PH3作为配体,使用半经验方法PM3(tm)并以从头算法和3-21G*基础改善结果,提出在23’中钯更接近于环丙基碳C1,相反的24’中过渡金属更接近于初级碳C3。25这些数据能够解释1(R=H)的取代反应(通过(软)亲核试剂)观察到的较高的反应活性和区域选择性。
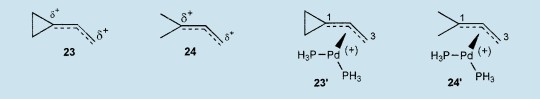
相反地,1(R=H)(从2e(R=H)或3b与Pd(dba)2/dppe制备出)与苯基锌氯化物25的反应分别以68%和75%的收率专门提供1-乙烯基-1-苯基环丙烷27。在初级碳中心上生成取代基,(2-苯基亚乙基)环丙烷28的不足使人想起与不稳定(硬)亲核试剂的反应遵循一个完全不同的机理。实际上,通过金属转移反应等和从锌到钯有机部分的传递,有机金属化合物反应生成σ-复合物26(环丙烷环上含有Pd),然后它经过Pd(0)还原消去反应产生27。7
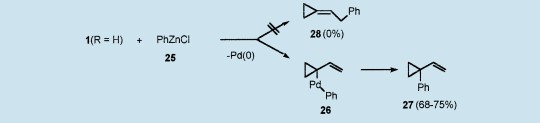
通过n-丁基锌氯化物,1(R=Ph)(来自1-苯乙烯基环丙基基本磺酸盐和Pd(dba)2,2PPh3)的取代反应以93%的收率仅生成1-苯乙烯基环丙烷31。氢解作用最可能在n-苯乙烯基取代σ-Pd复合物29(transmetalation)过程中出现,29经历β-消去反应形成σ-钯氢化物化学种类30,并在Pd(0)还原消去反应后生成31。据报道,丙烯基乙酸盐和含有β-氢的烷基基锌衍生物的Pd(0)催化反应通常产生还原产品,这是由于在少取代位上氢化物的攻击;26对1,1-二亚甲基丙烯基钯复合物还观察到相反的区域选择性。7,27,28

在[15]-冠(醚)-5存在时,通过甲酸钠,1(R=Bu)(来自1-(1-己烯基)环丙基甲苯磺酸盐和Pd(dba)2,2PPh3)的取代反应以80%的收率仅生成亚己基环丙烷33。27,28氢解作用很可能发生在甲酸σ-Pd复合物32经过氢化物的SNi传递的过程中。另外,1(R=Bu)与n-丁基锌氯化物的反应以85%的收率仅生成1-(1-己烯基)环丙烷。以同样的方式,通过甲酸钠,1(R=Ph)的氢解作用以81%的收率生成28.27,28

相应的1-(1-烯基)环烷基酯已经扩展了这些Pd(0)催化的还原反应;不仅通过环张力,而且通过磷配体的立体效应可以检测到区域选择性。它们可以为Wittig烯化作用提供一个简便的替换,特别是当底物对叶立德敏感时。27,28
在甲酸钠和[15]-冠(醚)-5的环境下,1(R=SiMe3)的取代反应以90%的收率仅生成(2-三甲基硅基亚乙基)环丙烷35,很可能通过σ-复合物34和SNi氢化物传递。相反地,很可能通过σ-复合物36和36’,1(R=SiMe3)与n-丁基锌氯化物的反应以85%的收率仅生成(2-三甲基硅基乙烯基)环丙烷37。28,29
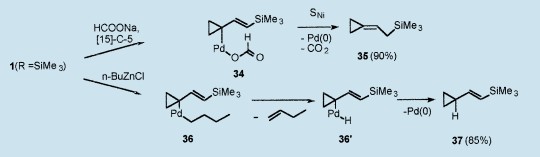
丙烯基硅烷和乙烯基硅烷构成了非常有用的多样合成中间体;30这些Pd(0)催化的还原反应(已经被扩大到相应的三烷基硅基丙烯基酯)提供一个新型,方便,区域选择性和立体选择性的方法以合成这些有用的亲电试剂。28,29
在胺,亚胺,羟胺,氨基甲酸酯类衍生物存在的环境下,1(R=H)的亲核取代反应生成了(2-环亚丙基乙基)胺(例如16-18),这是由少取代的丙烯基端的专一胺化(见上文)造成,并且从(2-环亚丙基乙基)酰亚胺38研究了环丙烷环的胺化反应,而38很容易从3a和三氯甲烷得到。31因此,在甲苯中简单的加热到100℃,38经历热诱导的aza-Claisen重排反应得到l-乙烯基环丙基酰亚胺39(96%的收率)。然后,39的氧化反应(NaIO4,RuCl3)和酰胺键的酸性裂解以87%的总收率生成l-氨基环丙烷羧酸(ACC)40,因为它的植物化学物质,酶,抗生素和神经化学活动,40目前吸引了特别的关注。2
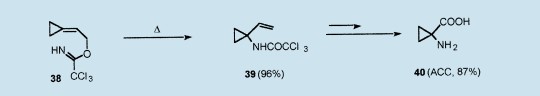
通常在过渡金属催化下也可以得到这样的热[3,3]迁移重排。然而与Pd(0)一起的38的处理没有像预期那样得到39;很明显,对于Pd(0)催化,亚胺盐38是惰性的,并且被未改变地回收。事实上,在这些环境下,很可能形成了1(R=H)和三氯乙酰胺阴离子41,但是由于氧气比氮气更为亲核,所以反应是可逆的,并重新生成了38。然而,如前文所说(见上),在2-钠丙二酸酯4存在的条件下,通过Pd(0)处理38定量生成5,毫无争议地证明了1(R=H)的出现。31
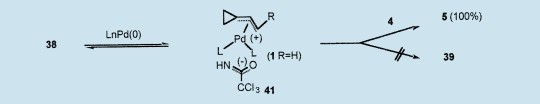
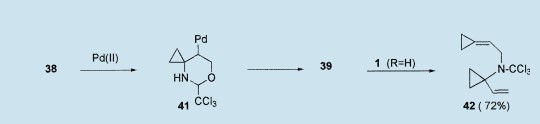
通过钯限制六元环阳碳离子中间体41,38与Pd(Ⅱ)催化剂一起的处理又引发了aza-Clasien反应并得到了预期的环丙基胺39。32但是,在原位也生成了复合物1(R=H)(很可能从41或直接从38生成),然后与39反应以72%的收率得到了意外地产物N-(2-环亚丙基乙基)-N-(1-乙烯基环丙基)三氯乙酰胺42。然而,为了避免随后的反应39¤42,预先对38进行N保护(例如像N-C6H4OMe),然后在Pd(Ⅱ)的催化下以74%的收率得到了39。31
在[15]-冠(醚)-5(10%)存在时,1(R=H,Ph)与叠氮化钠反应以79-80%的收率仅生成1-叠氮基-1-乙烯基环丙烷43(R=H,Ph),21而据报道,丙烯基乙酸盐20和1-乙烯基环己基乙酸盐经历Pd(0)催化的,带有相反区域选择性的叠氮化反应。33通过在甲醇中的1,3-丙烷二硫酚[HS(CH2)3SH]或氢氧化钠水溶液中三苯基膦的作用,43(R=H,Ph)的还原反应以60-79%的收率生成1-乙烯基环环丙基胺44(R=H,Ph)。44(R=Ph)中苯乙烯基的氧化裂解(NaIO4,RuCl3)也以73%的总收率得到氨基酸(ACC)40。21

从市场上可买到的旋光纯的(2S)-3-羟基-2-甲基丙酸酯制备出甲磺酸盐(1R,2S)-45(de:80%)。很可能通过复合物(2S)-46,它经过Pd(0)催化的立体选择胺化作用(NaN3,[15]-C-5),以85%的收率仅生成叠氮化合物(1R,2S)-48(de>92%)。34

丙烯基酯的Pd(0)催化胺化作用通常出现整体结构保持,33然而(2S)-46具有所需的构型转化的取代反应应该像前面所述,生成(环亚丙基乙基)叠氮化合物(2S)-47(软亲核行为,见上),而(2S)-46具有构型保持的取代反应反而应生成相应的(1S,2S)-叠氮化合物(硬亲核行为)。因此,(1R,2S)-48的专有生成反应必须是随后的Pd(0)诱导的(1R,2S)-47异构化的结果,通过钯部分协调双键立体控制异构化。34无论它可能是什么,(1R,2S)-48[HS(CH2)3SH]随后的消去反应和氧化裂解(NaIO4,RuCl3)方便地生成具有生物活性的氨基环丙烷羧酸(1R,2S)-49,并具有>99%的对映体过量。34
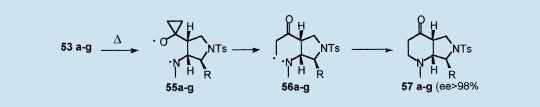
在NaH(1 equiv)存在的条件下,通过甲基N-甲苯磺酰基氨基酸酯50a-g,1(R=H)的取代反应以77-95%的收率生成甲基N-(2-环亚丙基乙基)-N-甲苯磺酰基氨基酸酯51a-g;对于从非对称的50b-g获得的51b-g(ee>98%),并没有观察到异构手性中心。酯51a-g[DIBALH(0.9equiv)或DIBALH(2.5equiv)和DMSO/(COCl)2]生成相应的醚,它在室温下与N-甲基羟基胺反应可生成N-甲基硝酮52a-g。这些硝酮52a-g以46-95%的收率自发地变成顺融三环异恶唑53a-g和54a-g。环亚丙基环张力(△S=40.9kcal/mol)与依赖于氨基酸部分上R取代基立体效应的非对映选择性一起,强烈地增加了这种区域选择性的分子内1,3-两极环加成反应的环加成率。36因此,R=Me太小不足以诱导53b和54b(de:10%)上的非对映选择性;更大的取代基R=i-Pr,PhCH2有利于54c,e(de:22-24%)的生成,然而R=3-吲哚基-CH2有利于53f(de:22%)的生成。另一方面,R=Ph和-(CH2)3-(甲基脯氨酸酯)生成纯非对映选择性环加合物53d,g(de>99%)。34通过分子机理计算(MAD),过渡态能量计算有效地证明,当R=Me(⊿E=1.5kcal·mol)时,外-R和内-R过渡态(可分别生成53a-g和54a-g)仅仅在能量上有细微的差别,但当R=Ph和脯氨酸(⊿E=4.39和10kcal·mol)时明显提高,所以毫无疑问的证明了这一非对映选择性。25,35

在二甲苯中简单加热到130-140℃,纯环加合物53a-g经过热诱导环增反应以冷人满意的收率得到吡咯[3,4-乙]-7-吡啶酮。经过弱N-O键的加热裂解,这种重排作用进行以生成环丙氧双自由基中间体5a-g,然后它很容易经过张力释放的环丙烷环开作用生成双自由基56a-g。然后在氮气和56a-g末端碳原子之间的分子内根耦合,以完整的化学、区域、非对映和对映选择性生成了旋光纯双环产物57a-g(ee>99%)。从纯环加合物54b,c,e观察到了相同的热行为。25,35当在硝酮生成后不需要附加试剂或催化剂时,这整个过程与最大化“原子经济”一起出现。24
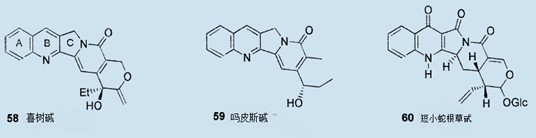
就在最近,作为P物质拮抗剂37或强力的DNA解旋酶抑制剂38,这种二氮杂二环[3,4]壬酮的衍生物显示了令人感兴趣的生物活性;它们作为主要疾病(哮喘、炎症、疼痛、偏头痛、血管性头痛)治疗的有前途的治疗药物出现。39此外,这种吡咯并[3,4]吡啶(或4-氮杂异吲哚)环系统存在于强力抗癌药物喜树碱5840、强力抗病毒药物吗皮斯碱5941和像短小蛇根草甙60那样相关天然产品中的B环、C环中,或者存在于非天然产品中。42
在NaH(1equiv)存在的条件下,1(R=H)与甲基羟乙酸酯61的反应以82%的收率生成了O-(2-环亚丙基乙基)羟乙酸酯62。同样的,62(DIBALH)的部分还原反应和N-甲基羟基胺的加成反应以90%的收率生成相应的硝酮,然后自发的变成呋喃[3,4-c]异恶唑63。这种三环类异恶唑63也经过热诱发环扩反应,以73%的收率生成呋喃[3,4-b]吡咯衍生物64。25从相应的环亚丙基烷基腈氧化物也已经成功获得这种简便的新工艺。25

另外,按完全一样的过程,在碱(LDA)存在时,在室温下五分钟内以87%的收率生成2-(2-环亚丙基乙基)甘氨酸衍生物66a表示N-(二苯基亚甲基)甘氨酸酯65a(X=OMe)作为亲核试剂使用。从碳酸盐2c(R=H),与希夫碱65a一起而没有其他附加的碱,该Pd(0)催化的C-甲基化作用发生,并以76%的收率生成66a。7,43樟脑磺内酰胺改性甘氨酸65b44(X=樟脑磺内酰胺)的使用以一个较好的非对映选择性(de>90%)生成66b。66b的脱保护生成旋光纯的(S)-2-氨基-4-环亚丙基酪酸67,正被恢复的手性辅助达90%以上的收率。43
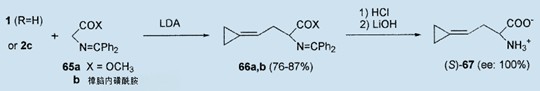
非天然化合物(S)-67是甲叉环丙基丙氨酸A[3-(2-亚甲基-环丙基)丙氨酸]68的一个同分异构体,68来自于西非荔枝果树()未成熟的果实,是牙买加呕吐病的病原体,并导致致命的低血糖和有机血症。45被认作是异基因-甘氨酸的(S)-67也抑制丙酮酸新陈代谢生成单糖,45但是并不能诱发脂肪酸的线粒体氧化。2

一种制备旋光纯α-丙烯基-α-氨基酸的一般简便的方法是基于这种手性改性甘氨酸等价物的钯催化烯丙基化。43
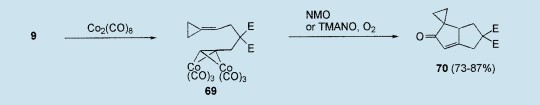
在最近五元碳环系统构建的方法中,炔烃的钴介导环加成反应以Pauson-Khand反应(PKR)而为人所知,反应中炔烃由于羰基插入而变成链烯烃,生成了一个环戊烯酮,这个反应已经引起了特别的兴趣。46在标准环境下,从八面羰基二钴[Co2(CO)8]和二甲基(2-环亚丙基乙基)炔丙基-丙二酸8开始制备烷基钴复合物69,其中8从1(R=H)(见上)中很容易获得。在氧环境下,69与N-甲基吗啡氮氧化物(NMO)或三甲基胺氮氧化物(TMANO)一起处理,以73和87%的收率分别地生成双环[3.3.0]辛烯酮70。与此相反,从22派生得到类似的二甲基2-(2-异亚丙基乙基)-2-炔丙二酸在相同的条件下并不环化,再一次说明,大环张力影响反应活性。47这一顺序被应用于种种不同取代的6-环亚丙基-1-己炔,以非常好的收率生成罗环丙烷双环[3.3.0]-1-辛烯-3-酮.47
70控制三奎烷倍半萜的基本框架是非常值得注意的,例如senoxydene 71, silphinene 72, a,b-cedrenes 73 和 modhephene 74。实际上,通过2-dioxanylethylcuprate简单的Michael加成反应已经改变了70,随后通过一个不对称缩合反应得到一个三奎烷71和72的合适前驱。48

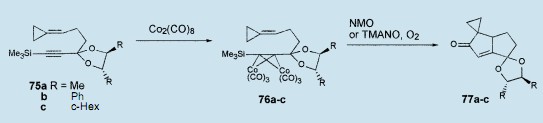
使用手性乙二醇衍生物,从1(R=H)制备不对称的1,6-烯炔75。虽然被大量取代(PKR对位阻现象非常敏感)46,75a-c经过促进环合作用的三炔胺氧化物(NMO或TMANO),并在钴复合物76a-c之后,以63,69,和76%的收率分别生成螺旋环丙烷双环[3.3.0]-1-辛烯-3-酮77a-c。对于77a(de:33%),当R=Me时,环合反应中的非对映选择性低;但对于77b(de:67%),当R=Ph时选择新增加;对于77c(de:73%),当R=环己基时选择性增加。47
与二甲基酮酸锂一起,经柱层析法分离的(S)-77b的得到进一步转变,在脱甲硅基(Me2CO,p-TsOH)后以85%的总收率生成双环[3.3.0]辛酮78,然后在延长加热后以59%的收率生成旋光纯的(5R)-二酮79。47,48

结合起来,容易获得亚烷基环丙烷的最方便的现行方法是基于通过软亲核试剂对复合物1的取代反应或通过以HCOONa为钠源的还原反应(见上)。显然,不对称的π-1,1-二亚甲基丙烯基钯复合物,比如以(2S)-46为例,能够生产光活性的亚烷基环丙烷。
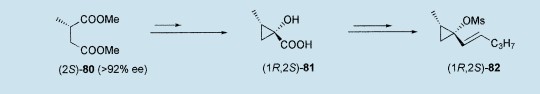
从它们的二甲基外消旋酸盐的酶(脂肪酶,酯酶)水解作用,49,50或手性酰亚胺烯醇化合物的立体选择烃化反应,51不对称的α-烷基琥珀酸盐在制备规模上是现成的。例如,(2S)-80(ee>92%)经过酮醇型环化作用(Na,CkSiMe3)和C4 ¤ C3环缩作用后,生成(1R,2S)-1-羟基-2-甲基环丙烷羧酸81,52作为甲磺酸盐(1R,2S)-82的合适前驱体来使用。12
通过甲酸钠和[15]-冠(醚)-5(见上),Pd(0)催化的(1R,2S)-82氢解反应以高收率(79-85%)生成旋光纯的(E)-(2S)-亚戊烷基(2-甲基环丙烷)84和(1R,2S)-2-甲基-1-(1-戊烯基)环丙烷85的分离混合物,该反应有被配体性质大大影响的区域选择性。事实上,三价磷配体的立体效应以控制过渡金属复合物的化学行为而为人所知。53就像对复合物1(R=Ph,Bu)先期观察到的,27,28当作为钠源使用HCOONa时,磷上取代基尺寸的增加有利于亚烷基环丙烷(2S)-84的形成,而当n-丁基锌氯化物是氢源时,尺寸增加不利于乙烯基环丙烷衍生物(1R,2S)-85的形成。因此PPh3,P(P-甲氧基)3和P(0-甲氧基)3和P(o-甲苯基)的成功使用把(2S)-84/(1R,2S)-85比率从63/37增加到81/19。12

然后,通过m-氯per苯甲酸(MCPBA),(2S)-84(de:100%)环氧化反应以90%的收率生成(2R,3S,4S)-86和(2S,3R,4S)-87的7:3混合物,这构成了不对称的氧杂螺戊烷的第一个全合成。54

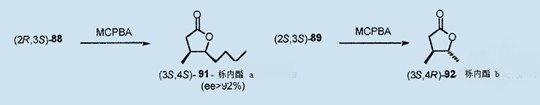
在用催化量的碘化锂(1%)处理上,环氧作用的混合物经过定量的C3 ¤ C4环扩作用后,生成非对映的(2R,3S)-88,(2S,3S)-2-丁基-3-甲基环丁酮89以及区域异构体2-丁基-4-甲基环丁酮90的55:17:28的混合物。

用MCPBA进一步处理分离出的环丁酮(2R,3S)-88,生成(3S,4R)-4-丁基-3-甲基丙酮酰91(ee>92%);而在相同的环境下,(2S,3S)-89变成非对映的内酯(3S,4S)-92(ee>89%)。54从白橡木可分离出这两种丁酮酰(以Quercus内酯a和b为人所知),而且储存在橡木桶中的红酒和烈酒中发现了这两种丁酮酰。54
这些丁酮酰的第一次非对称合成涉及1-乙烯基环丙醇衍生物的酸诱导环扩反应,也从α-羟基酸(1R,2S)-81和琥珀酸盐(2S)-80中生成。52非对称的环丁基酮(例如(2R,3S)-88 和 (2S,3S)-90)现在不仅可容易地用来向γ-丁内酯(MCPBA)提供合适的前驱,而且向环丙酮(CH2N2)和γ-丁内酰胺(ArSO2ONH2)提供。56
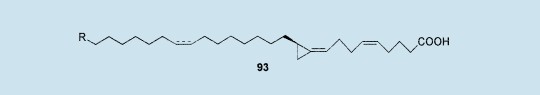
最近,从澳大利亚海洋海绵属amphimedon中已经分离出amphimic酸93(R=H,Me)。作为DNA拓扑异构酶Ⅰ抑制剂,这种紧脂肪酸(C27-C28)(拥有一个环亚丙基部分(SE:40.9kcal·mol-1))的活性比链C18高100倍,而且也显示了对抗P388白血细胞(IC50:1.8μm)的细胞毒性。57合成涉及像1这样的π-1,1-二亚甲基钯复合物,现在它们的全不对称合成正在调查研究中。58
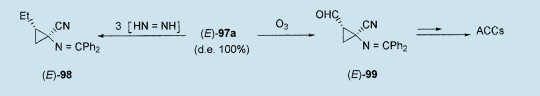
最近提出制备高功能化环丙基(E)-97a-b的替代方法,即通过Pd(0)催化的一锅串联烷基化和通过不同α取代的乙腈95a-d1,4-二环丁-2-烯(市场上可以买到的)的SN’环化。通过两性离子中间体π-丙烯基钯复合物96a-d,该反应发生并以67-87%的收率生成2-乙烯基-1-取代的环丙烷腈(E)-97a-d。96a-d的SN’环化具有较高的非对映选择性(de:88-100%);通过π-丙烯基钯部分地攻击和负碳离子的构型转变(涉及乙烯基和腈基团的立体青睐原位关系),该反应发生。59

例如,(E)-97a(de:100%)的简单二酰亚胺还原生成(E)-98,它作为合适的coronamic酸的前驱体使用。59,60另外,臭氧分解能够生成具有生物价值的环丙烷甲醛(E)-99,2也能生成2,3-环己甲醇氨基酸(ACCs)61。
有人试图通过这种新前驱体的方法得到非对称合成,使用手性钯配体[例如(S)-或(R)-BINAP],手性氨基乙腈[来自1-羟基蒎烷酮]或手性丙烯基氯化物[来自(2S)-甲基乳酸盐],已经生成了(1S,2R)-97a及其衍生物,这个反应具有完全的非对映选择性(de=100%),但对映选择性很低(ee≤32%)。61事实上,这些结果已经指出了环化步骤96a¤97a的可逆性,以及Pd(0)诱导的乙烯基环丙烷温和环开化的结果。62为了解决这个问题,通过希夫碱95a阴离子,从丙烯基氯化物(4S)-100[来自(2S)-甲基乳酸盐,ee>94%]的Pd(0)催化的烷基胺化反应,以97%的产率制备2-氨基-6-羟基庚-4-烯腈(6S)-100。当钯存在,但在Mitsunobu反应环境(DEAD/PMe3)下,(6S)-101经过预期的非对映选择的环化作用,以77%的产率生成1-氨基环丙烷-腈衍生物(1S,2R)-102(ee>83%),它也构成了ACCs的一个有效地前驱体。61

由此可得出结论,在1-(1-烯基)环丙酯2b-g或3b,c上存在烯丙基离去基团时,(1R,2S)-45和(1R,2S)-82对于阻止三元环开和致使π-1,1-二亚甲基丙烯基钯复合物(例如1,(2S)-46和(1S)-83)生成的不可逆是至关重要的。
参考文献
1. For recent books, see: Carbocyclic Three- and Four-membered Ring Systems. Methods of Organic Chemistry; de Meijere, A., Ed.; Houben-Weyl; Thieme Stuttgart: New York, 1997; Vol. E l7a-f.
2. Salaün, J.; Baird, M. S. Curr. Med. Chem. 1995, 2, 511; Salaün, J. Top. Curr. Chem. 1999, in press.
3. Liu, H.-W.; Walsh, C. T. The Biochemistry of the Cyclopropyl Group in The Chemistry of Functional Groups; Patai, S., Ed.; Wiley: New York, 1987; Vol. 2, p 259.
4. Reissig, H.-U. Top. Curr. Chem. 1988, 144, 73.
5. Salaün, J. Top. Curr. Chem. 1988, 144, 1.
6. Schnaubelt, J.; Ulmann, A.; Reissig, H.-U. Synlett 1995, 1223; Ulmann, A.; Reissig, H.-U.; Rademacher, O. Eur. J. Org. Chem. 1998, 2541.
7. Stolle, A.; Ollivier, J.; Piras, P. P.; Salaün, J.; de Meijere, A. J. Am. Chem. Soc. 1992, 114, 4051; Stolle, A.; Salaün, J.; de Meijere, A. Tetrahedron Lett. 1990, 31, 4593.
8. Salaün, J.; Conia, J. M. Tetrahedron Lett. 1972, 2849; Salaün, J.; Garnier, B.; Conia, J. M.Tetrahedron 1974, 30, 1413.
9. Salaün, J. Chem. Rev. 1983, 83, 619; Salaün, J. Encyclopedia Reag. Org. Synth., 1994, Vol. 4, p 2358.
10. Salaün, J.; Almirantis, Y. Tetrahedron, 1983, 39, 2421; Salaün, J. Encyclopedia Reag. Org. Synth., 1994, Vol. 7, p 4773.
11. Kulinkovich, O. G.; Sviridov, S. V.; Vasilevski, D. A. Synthesis 1991, 234; Kulinkovich, O. G.; Sviridov, S. V.; Vasilevski, D. A.; Prityckaja, T. S. Zh. Org. Khim. 1989, 25, 2245.
12. Chevtchouk, T.; Ollivier, J.; Salaün, J. Tetrahedron: Asymmetry 1997, 8, 1005; Lee, J.; Kim, H.; Cha, J. K. J. Am. Chem. Soc. 1996, 118, 4198; Kasatkin A.; Sato, F. Tetrahedron Lett. 1995, 36, 6079.
13. Atlan, V.; Racouchot, S.; Rubin, M.; Bremer, C.; Ollivier, J.; de Meijere, A.; Salaün, J. Tetrahedron: Asymmetry 1998, 9, 1131.
14. Moss, R. A.; Munjal, R. C. Synthesis 1979, 425.
15. Liese, T.; Jaekel, F.; de Meijere, A. Org. Synth. 1990, 69, 144.
16. Spitzner, S.; Swoboda, H. Tetrahedron Lett. 1986, 27, 1281.
17. All these precursors of esters 2b-g and 3b,c are commercially available.
18. Tsuji, J.; Minami, I. Acc. Chem. Res. 1987, 20, 140; Trost, B. M. Acc. Chem. Res. 1990, 23, 34.
19. Tsuji, J.; Shimizu, I.; Minami, I.; Ohashi, Y.; Takahashi, K. J. Org. Chem. 1985, 50, 1523.
20. Mc Gaffin, G.; Michalski, S.; Stolle, A.; Bräse, S.; Salaün, J.; de Meijere, A. Synlett 1992, 558.
21. Aufranc, P.; Ollivier, J.; Stolle, A.; Bremer, C.; Es-Sayed, M.; de Meijere, A.; Salaün, J. Tetrahedron Lett. 1993, 34, 4193.
22. For a review see: Goti, A.; Cordero, F. M.; Brandi, A. Top. Curr. Chem. 1996, 178, 1.
23. For a review see: Brandi, A.; Goti, A. Chem. Rev. 1998, 98.
24. Trost, B. M. Angew. Chem. Int. Ed. Engl. 1995, 34, 259.
25. Ferrara, M.; Cordero, F. M.; Goti, A.; Brandi, A.; Estieu, K.; Paugam, R.; Ollivier, J.; Salaün, J. Eur. J. Org. Chem. 1999, in press.
26. Matsushita, H.; Negishi, E. J. Org. Chem. 1982, 47, 4161.
27. Ollivier, J.; Piras, P. P.; Stolle, A.; Aufranc, P.; de Meijere, A.; Salaün, J. Tetrahedron Lett. 1992, 33, 3307.
28. Ollivier, J.; Dorizon, Ph.; Piras, P. P.; de Meijere, A.; Salaün, J. Inorg. Chim. Acta 1994, 222, 37.
29. Ollivier, J.; Salaün, J. Synlett 1994, 949.
30. For a review see: Fleming, I.; Dunogues, J.; Smithers, R. Org. React. 1989, 37, 57.
31. Estieu, K.; Ollivier, J.; Sa1aün, J. Tetrahedron Lett. 1995, 36, 2975.
32. Schenck, T. G.; Bosnich, B. J. Am. Chem, Soc. 1985, 107, 2058.
33. Murahashi, S.; Taniguchi, Y.; Imada, Y.; Tanigawa, Y. J. Org. Chem. 1989, 54, 3292.
34. Atlan, V.; Racouchot, S.; Rubin, M.; Bremer, C.; Ollivier, J.; de Meijere, A.; Salaün, J. Tetrahedron: Asymmetry 1998, 9, 1131.
35. Estieu, K.; Paugam, R.; Ollivier, J.; Salaün, J.; Cordero, F. M.; Goti, A.; Brandi, A. J. Org. Chem. 1997, 62, 8276.
36. Aurich, H. G.; Frenzen, G.; Gentes, C. Chem. Ber. 1993, 126, 787.
37. Fardin, V.; Foucault, F.; Bock, M. D.; Jolly, A.; Flamand, O.; Clerc, F.; Garrett, C. Neuropeptides 1994, 26, 34.
38. Qun, L.; Chu, D. T. W.; Clairborne, A.; Cooper, C. S.; Lee, C. M.; Raye, K.; Berst, K. B.; Donner,P.; Wang, W.; Hasvold, L.; Fung, A.; Ma, Z.; Tufano, M.; Flamm, R.; Shen, L. L.; Baranowski, J.; Nilius, A.; Alder, J.; Meulbreek, J.; Marsh, K.; Crowell, D.; Hui, Y.; Seif, L.; Melcher, L. M.; Henry, R.; Spanton, S.; Faghih, R.; Klein, L. L.; Tanaka, S. K.; Plattner, J. J. J. Med. Chem. 1996, 39, 3070.
39. Peyronel, J. F.; Tabart, M.; Achard, D.; Malleron, J. L.; Grisoni, S.; Carruette, A.; Montier, F.; Moussaoui, S.; Fardin, V.; Garrett, C. Eur. J. Med. Chem. 1995, 30 (suppl.), 5765.
40. Josien, H.; Ko, S. B.; Ban, D.; Curran, D. P. Chem. Eur. J. 1998, 4, 67.
41. Josien, H.; Curran, D. P. Tetrahedron 1997, 53, 8881.
42. Henegar, K. E.; Ashford, S. W.; Baughman, T. A.; Sih, J. C.; Gu, R. L. J. Org. Chem. 1997, 62, 6588.
43. Voigt, K.; Stolle, A.; Salaün, J.; de Meijere, A. Synlett 1995, 226.
44. Josien, H.; Lavielle, S.; Brunissen, A.; Saffroy, M.; Torrens, Y.; Beaujouan, J. C.; Glowinski, T.; Chassaing, G. J. Med. Chem. 1994, 37, 1586.
45. Sherrat, H. S. A. Trends in Pharmac. Sc. 1986, 186; Billington, D.; Osmundsen, H.; Sherratt, H. S. A. Biochem. Pharmacol. 1979, 27, 2891.
46. For a review see: Schore, N. E. Org. React. 1991, 40, 1.
47. Stolle, A.; Becker, H.; Salaün, J.; de Meijere, A. Tetrahedron Lett. 1994, 35, 3517 and 3521.
48. Stolle, A. Thesis, Hamburg 1992.
49. Guibé-Jampel, E.; Rousseau, G.; Salaün, J. J. Chem. Soc., Chem. Commun. 1987, 1081.
50. Chevtchouk, T.; Ollivier, J.; Salaün, J.; Merlet, D.; Courtieu, J. Tetrahedron: Asymmetry 1997, 8, 999.
51. Salaün, J.; Fadel, A. Tetrahedron Lett. 1988, 29, 6257.
52. Salaün, J.; Karkour, B.; Ollivier, J. Tetrahedron 1989, 45, 3151; Salaün, J.; Karkour, B. Tetrahedron Lett. 1988, 1537.
53. Tolman, C. A. Chem. Rev. 1977, 77, 313.
54. Chevtchouk, T.; Ollivier, J.; Salaün, J. Tetrahedron: Asymmetry 1997, 8, 1005 and 1011.
55. Günther, C.; Mosandl, A. Liebigs Ann. Chem. 1986, 212.
56. Delair, Ph.; Kanazawa, A. M.; de Azevedo, M. B. M.; Greene, A. E. Tetrahedron: Asymmetty 1996, 7, 2707.
57. Nemoto, T.; Ojika, M.; Sakagami, Y. Tetrahedron Lett. 1997, 38, 5667; Nemoto, T.; Yoshino, G.; Ojika, M.; Sakagami, Y. Tetrahedron 1997, 53, 16699.
58. Appolonova, S.; Racouchot, S.; Ollivier, J.; Salaün, J., unpublished results.
59. Gaucher, A.; Dorizon, Ph.; Ollivier, J.; Salaün, J. Tetrahedron Lett. 1995, 36, 2979 ; Franzone, G.; Carle, S.; Dorizon Ph.; Ollivier, J.; Salaün, J. Synlett 1996, 1067.
60. Gaucher, A.; Ollivier, J.; Marguerite, J.; Paugam, R.; Salaün, J. Can. J. Chem. 1994, 72, 1312.
61. Dorizon, Ph.; Su, G.; Ludvig, G.; Nikitina, L.; Ollivier, J.; Salaün, J. Synlett 1998, 483; Dorizon, Ph.; Su, G.; Ludvig, G.; Nikitina, L.; Paugam, R.; Ollivier, J.; Salaün, J. J. Org. Chem. 1999, in press.
62. Yamamoto, K.; Ishida, T.; Tsuji, J. Chem. Lett. 1987, 1157; Burgess, K. Tetrahedron Lett. 1985, 26, 3049; Larock, R. C.; Yum, E. K. Synlett 1990, 529.
注:本文为提供者翻译的,由于知识所限,其中错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号