用于对映体拆分、绝对构型的测定和质谱测定对映体过量的新型强效手性酸
- 2012-06-04
- 专题
1.前言
现在大家一致认为分子的手性是生命过程中必不可少的部分,还一致同意大多数控制活生物体生理机能的生物活性化合物都是具有手性的。因而,在针对生物活性化合物以及天然产物的结构研究上,其绝对构型的测定成为首要的重大问题。其次是成为医药目标的生物活性化合物和天然产物的手性合成,以及如何有效的利用100%对映体纯度和对映体过量(% ee)来合成所需的对映体。更深层次的讲是研究手性功能分子和分子机器,例如我们实验室开发研制的光动力手性分子马达在近几年发展比较迅速。因此,在材料科学领域,手性分子绝对构型的准确测定与绝对构型在手性合成中的应用一样极其重要。
我们最近开发研制的手性羧酸作为新型手性助剂,已被证明是对映体拆分强有力的分子工具并且能明确的测定各种醇的绝对构型。这些手性酸对于采用100%对映体过量简便合成对映体分子是非常有用的,对其绝对构型的分配也有着重要的作用。采用这些手性羧酸的各种合成方法已被成功的应用于各种化合物的合成,本文稿详尽地解释了其基本合成方法和应用。
2.绝对构型的确定方法及评价
确定手性化合物的绝对构型的方法主要分为以下两类:
(1)确定手性化合物绝对构型的非经验方法
该类别中的方法包括通过X射线晶体学的Bijvoet手性方法1)和圆二色谱激子手性方法2)。这两种功能强大的方法可以在无需知道相应的参考化合物绝对构型的前提下提供目标分子构型的非经验测定。对于X射线晶体学,因为重原子的不规则色散效应在适当条件下可以得到非常准确的测定,所以我们可以依此获得清晰而明确的绝对立体结构。因为分子可以采用三维立体结构的形式被投射出来以至于该方法如今被广泛的采用。不过,X射线方法需要一个合适大小的单一晶体来适用于X射线衍射,所以此方法的关键问题在于如何获得这样的单晶。因此,采用这种方法的研究通常会变成一个漫长的试验以及对于一种理想单晶的错误搜寻。
圆二色谱激子手性方法2)与前一个方法同样的重要,是因为绝对构型可以采用非经验方法确定并且不需要结晶过程。再者可用圆二色谱追踪手性化学反应和生物反应,甚至还可以采用这种方法来获得不稳定化合物的绝对构型和构象。然而,由于一些化合物并不是适用于这种方法的理想对象,所以所获得结果必须仔细地加以解释。
(2)采用已知构型化合物作内参比测定绝对构型的相对方法
通过以已知绝对构型的一个参比化合物或一个取代基作对比来测定感兴趣位点上的相对构型,从而可以获得目标化合物的绝对构型。一个典型的例子就是向试样中引入已知绝对构型的手性辅助剂后再采用X射线晶体学确定其绝对构型3-6)。在这种情况下,根据所引入作为内参比物辅助剂的手性就可以自动确定有问题的这一点的绝对构型。因而,所用样品不需要包含有带反色散效应的重原子。
甚至因单晶质量低劣而引起最终的R值不是足够小,我们也能获得非常清晰的结果。即便仅仅获得了相对构型也可以确切的确定其绝对构型。目前已经形成了很多种将内参比物连接到目标分子上的方法,例如,有离子键诸如传统的酸碱盐类,共价键诸如酯类或酰胺类以及近来研究开发的包合物的使用7-9)。这类相对的X射线方法有望得到更为广泛的应用。
近年来,氢质子核磁共振(1H NMR)各向异性方法已经经常被用作相应的方法来确定绝对构型,同时在研究天然产物的绝对构型上有着重要的作用。尤其是经常采用Kusumi 等人10-13)开发的改进Mosher方法确定仲醇的绝对构型。如此看来,如果已知手性辅助剂[Mosher试剂α-甲氧基-α-(三氟甲基)苯乙酸(MTPA)]和[Trost试剂α-甲氧基苯乙酸]的绝对构型,那么就可以自然而然的知道由手性仲醇与α-甲氧基-α-(三氟甲基)苯乙酸或α-甲氧基苯乙酸反应生成的酯的绝对构型。再者,由于在外部磁场的诱导下产生环电流致使芳香取代基(苯基)产生磁各向异性效应,朝着优势构象中苯基的醇基部分的核磁共振信号移动到一个更高的磁场(称为高场移)。通过观察氢质子核磁共振各向异性效应来确定醇组分的绝对构型。由于其不需要化合物结晶且核磁共振机器很容易测定所以这种方法非常方便,不过此方法的一个缺点是它是基于在溶液中分子的优势构象的假设,而优点是测定非常可靠因为此方法本身有着自诊断功能。该方法已广泛地应用于仲醇构型的测定,更有望扩展到其他类型化合物构型的测定上。
绝对构型的相对确定还可以采用化学相关法,光学旋光[α]D比较法或者已知绝对构型参考物的圆二色谱法。虽然这种方法也被经常采用,但是仔细挑选参考物对于一个可靠分析来说是非常必要的。

3.手性合成方法及其评价
确定了手性化合物绝对构型后的主要任务就是要合成该手性化合物。合成手性化合物切实可行的方法大致可以分为两类,且每一类还可再分;这些合成方法的优缺点如下文所述。本文中手性合成主要包括:所谓的不对称合成和对映体拆分。此外,该方法首先采用手性辅助剂形成一个共价键合的非对映异构体,然后由高效液相色谱分离最后将目标化合物修复,在广泛意义上也被定义为对映拆分。
(1)外消旋体的对映拆分
类型a). 在这个方法中,手性辅助剂先离子键合到外消旋体上形成非对映异构体混合物,然后经分步重结晶获得对映异构体化合物。这种方法也适用于例如氢键结合7-9)的包合物的形成,关键点是能否用100%对映体纯度通过分步重结晶来获得非对映体。值得注意的是重结晶并非经常能获得100%对映纯的非对映体,如果此方法成功了,它便适合大规模的制备手性化合物。
类型b). 在该方法中手性辅助剂先共价键合到外消旋体上生成非对映体混合物,混合物再经硅胶高效液相色谱或其他方法分离出成对映纯非对映异构体,然后手性辅助剂裂解下来(图1)。该方法也能产生一种对映体化合物,问题是能否通过高效液相色谱把这些非对映异构体有效地分离,如果可以有效的分离开那么所获得每一种非对映体都是对映纯化合物,手性辅助剂裂解后所得到的目标化合物也是100%对映纯。最好的做法是采用一个能够轻松裂解的手性辅助剂。
类型c). 此方法非常好地其采用了带有手性柱(以手性固定相制成的色谱柱)的高效液相色谱或气相色谱直接对外消旋体进行对映体分离,这类已经有很多研究报道过了14)。其关键问题也是能否将外消旋体分离成两种对映体,如果能有效的分离那么可以通过此方法获得100%纯的对映体。该方法比较简便适合用于分析分离,特别是因为它不需要衍生过程。总的来说手性柱都比较昂贵,大多用于分析,不过在一些情况下,工业上获得手性化合物大多采用大规模分离如医药分离等。通过洗脱顺序来确定绝对结构时必须仔细分析,因为会有不少异常情况出现。
类型d). 此方法比较特别,因为外消旋体经过酶或不对称反应的动力学拆分效应产生对映体。特别是酶反应的高立体选择性可以产生高对映体纯度对映体15),然而应该小心的是该方法并不是总产生100%对映体纯度。
(2)不对称合成
类型a). 这是一个高效的、功能性强的合成方法,该方法通过手性试剂或手性催化剂在非手性化合物上反应来获得手性产物。作为一个众所周知的合成方法,许多著名的综述报道了这些不对称合成,也没有必要再做进一步的解释。但是,该方法的缺点是其获得的产物并不都是对映纯。进一步讲,若是基于反应机理便很难确定产物的绝对构型。因此,我们建议采用以上描述的方法来独立地确定绝对构型。
类型b). 还有另外一种获得手性化合物的方法例如通过非手性或内消旋化合物上的酶反应生成手性化合物。通过酶催化内消旋化合物的不对称合成反应尤其有趣,并被定义为去对称化反应。在此情况下,对映体纯度也不能达到100%,必须分别确定绝对构型。
4.用于高效液相色谱对映拆分与X-射线晶体学的强功能手性辅助剂:羧酸的应用
实验室制备适量的具有100%对映体纯度的各种手性化合物,对映拆分1b)被认为是一种高效的方法,如图1所示。本方法中一种手性辅助剂共价键合到外消旋体上生成非对映体混合物,然后再经硅胶高效液相色谱分离。如果色谱图上显示了基线分离,说明所获得的非对映体是对映纯的。该方法的特点是即使在少量情况下也能清晰、有效地分离,类型1a)描述了其与分步重结晶的对比。
我们用硅胶高效液相色谱实验了含氮非对映体化合物(如腙16)、酰胺衍生物17))的分离。硅胶高效液相色谱能非常有效地分离手性胺与外消旋羧酸反应生成的酰胺类非对映异构体。然而,由于酰胺键不容易水解,羧酸的回收是要解决的问题。因此,需要一些额外的反应如水解前的亚硝化反应等17)。
我们还发现(1S,2R,4R)-(-)樟脑磺内酰胺(5)作为一种手性辅助剂被成功地应用在不对称合成上,对各种羧酸的对映拆分反应也至关重要。所获得的樟脑磺内酰胺类非对映体混合物能很好的用硅胶高效液相色谱进行分离,再者分离后的非对映体樟脑磺内酰胺键能立即被LiAlH4还原裂解生成伯醇,随后通过氧化迅速转化成羧酸对映体。
我们进一步发现由2,10-樟脑磺内酰胺(5)制得的酰胺一般都有良好的结晶度,很可能能够产生X-射线晶体学所必需的大单晶3-6)。一般来说,需要进行一系列的实验来获得能够立即产生单晶的衍生物,通过此对映拆分方法可以同时获得两种非对映体,也提供了两种生成单晶的可能性。如果能成功的用X-射线分析一种非对映体,那么另外一种非对映体的绝对构型也就自然而然的确定了。
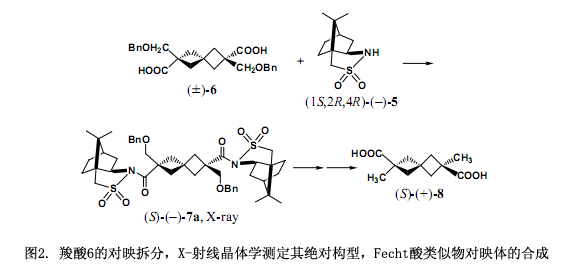
由于手性辅助剂(1S,2R,4R)-(-)2,10-樟脑磺内酰胺(5)(TCI,东京化成工业株式会社)是由天然的(1R,4R)-(+)-樟脑油18)合成而来,所以已经知道了它的绝对构型。因而,它可以用来作为一个通过X-射线晶体学确定绝对构型的内参比物,也可以确定羧基部分的绝对构型。2,10-樟脑磺内酰胺(5)含有一个硫原子作为重原子,使它能够采用Bijvoet方法来确定其绝对构型。因此,可以采用两种方法分别确定其绝对构型。
一个典型的例子如图2所示3),(1S,2R,4R)-(-)2,10-樟脑磺内酰胺(5)用NaH处理后生成一个阴离子,然后与酸性氯化物(±)-2,6-二(苄氧基甲基)螺[3,3]庚烷-2,6-二羧酸(6)反应,生成两个非常容易分离的非对映体7a和7b,并在5cm的薄层色谱硅胶板上出现两个明显的黑斑(∆Rf = 0.12)。在我们的实验中单用硅胶(22φ × 300 mm: 苯/乙酸乙酯= 20:1): Rs =2.9高效液相色谱分离了大约100mg。
第一部分(–)-7a通过乙酸乙酯的重结晶产生柱状晶体,然后采用X-射线晶体学分析单晶,使用樟脑部分作为一个内参比物明确地测定其绝对构型为S。此外,由重原子反常色散效应所确定的绝对构型与内参比方法所测定的完全一致。采用LiAlH4还原(S)-(–)-7a断裂掉手性辅助剂后,经过几步反应转变成(S)-(+)-2,6 -二甲基螺[3.3]庚烷-2,6-二元酸(8)。这样,我们合成了非消旋体和Fecht酸类似物8的对映体,并确定了其绝对构型3)。
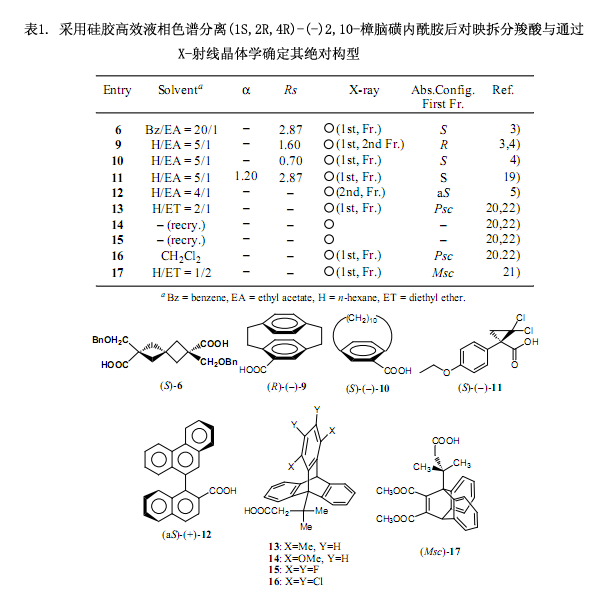
其它的应用例子如图1所示,该方法应用于具有不同结构的多种羧酸的对映拆分,因为其显示出较好的结晶度,所以用X-射线分析能很容易确定其绝对构型。个别化合物绝对构型的研究在表1中列出,并揭示了一些有趣的事实。例如,曾经通过与已知绝对构型化合物的化学相关性确定[8] 二聚二甲苯类-10-羧酸的绝对构型为(S)-(+)型。然而,用我们的方法4)证明此种构型是错误的,我们得出的结论是化合物9和10的构型也应该加以修订。尽管采用化学相关性所确定的绝对构型被认为是最可靠的方法,但是像这样的错误很容易发生,所以在使用时应多加注意。
由Toyota等人研究的化合物13-17也是具有旋转对映异构性和光学活性物质的有趣例子。总的来说,基于空间位阻旋转对映异构现象很难确定手性化合物的绝对构型,不过Toyota等人采用上述方法成功的解决了这个问题20-22)。
5.用于高效液相色谱法和X-射线晶体法分析醇类对映拆分的高效手性辅助剂的分子设计
上文已经详细叙述的手性助剂2,10-樟脑磺内酰胺(5)在对映拆分以及羧酸绝对构型的确定上至关重要,然而,对于手性醇合成以及确定其绝对构型的需求远大于羧酸。关键的问题是在保留上述手性助剂特点的同时要采用什么样的助剂来适用于这类醇。
我们采用以下的分子设计来处理这个问题,首先,设计引入了连接2,10-樟脑磺内酰胺和醇的连接物(图3)23),2,10-樟脑磺内酰胺的一个酰胺键和醇的一个酯键选择性键合到连接物上,由于酰胺键跟硅胶高效液相色谱亲和力较强而被保留下来,而酯键很容易转变以及裂解掉常被用来生成醇。因此,邻苯二甲酸作为连接分子的首选23),在对苯二甲酸和琥珀酸上两个手性部分在空间上被分割开来。但是,在邻苯二甲酸上两个手性部分比较接近以至于产生更强的相互作用。我们推断高效液相色谱能有效地识别其非对映体。
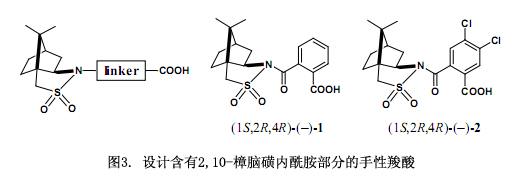
目标物手性邻苯二甲酸(–)-1是由(1S,2R,4R)-(-)2,10-樟脑磺内酰胺(5)阴离子与邻苯二甲酸酐反应所合成的,在CHCl3中的熔点为184-187℃;[α]D20 -134.7 (c 2.218, 甲醇)。化合物1应正式称为手性邻苯二甲酸酰胺,不过在这里我们采用其通用名手性邻苯二甲酸。在二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP)存在条件下,用醇来缩合该羧酸23)。
以下例证详述了制备步骤,手性邻苯二甲酸(-)-1可以与(±)-α-甲基-(4 - 溴苄基)甲醇(18)(表2所示)反应,随后用硅胶高效液相色谱(正己烷/乙酸乙酯= 3:1; α = 1.1, Rs = 1.3.)分离所生成的非对映体混合物。如果分离因数α大于等于1.1,那么所生成的化学组分通常是可以完全分离。酯19b作为第二组分被洗脱出来而且具有良好的结晶度,经过在甲醇溶液中的重结晶后生成适合于X-射线分析的单晶。从X-射线扫描的ORTEP图上看出,通过采用2,10-樟脑磺内酰胺部分作为内部参考物以及采用重原子效应测定醇部分的绝对构型,可以清晰地将其确定为S型。还从非对映体酯19b中重新获得了对映体醇(S)-(–)-1823)。
如表2所示的其他例子,我们成功的对映拆分了各种醇并采用X-射线分析确定了其绝对构型,表中所列举回收的对映纯醇再次证明了此方法在合成上的有效性。在氰醇27和胺28中可以采用这种方法进行非对映体分离和绝对构型的确定。不过,存在的问题是对映纯化合物27与28如何回收,原因是酰胺键不容易水解掉。酰胺28作为(2R,3R)-(+)-酒石酸19)的盐被对映分解,通过采用此方法确定了其绝对构型是(S)-(–)型。对于化合物29即使在它的结晶度较差所导致R值比较小的情况下,也可以通过采用内参比方法清晰的确定其绝对构型。
在我们研究的过程中,我们发现手性邻苯二甲酸(–)-1的酯类通常具有较差的溶解性,可能是因为其极易结晶而导致其在硅胶高效液相色谱里洗脱时间延长。再者就是在重结晶过程中生成细微针形的晶粒致使其不适合用X-射线晶体学分析。这说明使用低溶解性的手性邻苯二甲酸酯具有一定的反作用,因此我们在探索一系列的化合物中应该寻找能生成具有较强溶解性的软酯连接物。
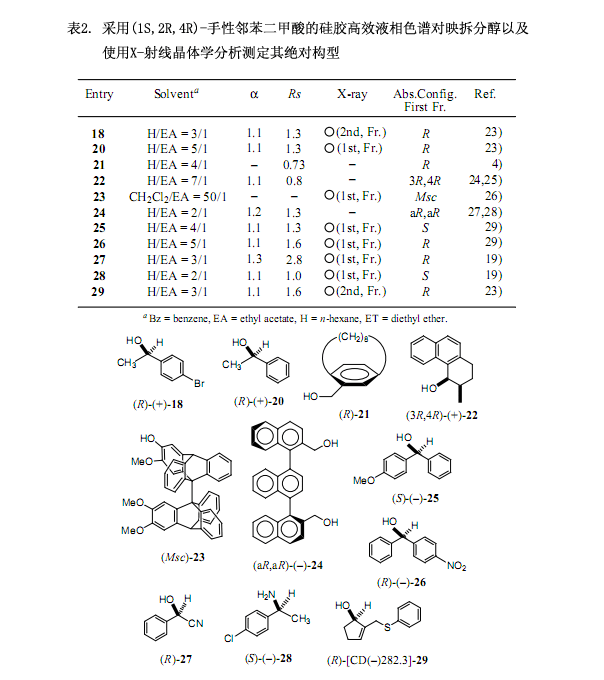
6.较强功能手性二氯代邻苯二甲酸用于醇的高效液相色谱对映拆分和X-射线晶体分析:发展与应用
经过一系列的调查研究,我们发现手性二氯代邻苯二甲酸(–)-2[在乙醇中的熔点221℃;[α]D20 –101.1 ;(c 1.375, 甲醇);图3]是由4,5-二氯代邻苯二甲酸作连接物而制备,有效地解决了上述讨论的问题,也就是其具有较高的溶解性能和在高效液相色谱中的洗脱时间比较短。并且,还能提供适用于X-射线分析的柱状晶体。例如对于表2及图4中的(±)-顺式-1,2,3,4 -四氢-3-甲基-4-菲酚(22)来说,采用手性邻苯二甲酸洗脱这两个非对映体时间分别为27.6分钟和31.0分钟,但是在同样的条件下采用手性二氯代邻苯二甲酸洗脱的时间分别是14.6和16.7分钟,后者所用的时间是前者的一半左右,更进一步说就是提高了其分离和分配因子24)。
手性辅助剂二氯代邻苯二甲酸(–)-2是一种重要的内参比物在采用X-射线分析测定绝对构型时,跟手性邻苯二甲酸(–)-1作用相似。其中,羧酸2除了含有一个硫原子外还含有2个氯原子作为重原子,因而通过重原子的反常色散效应可以有效地测定绝对构型。
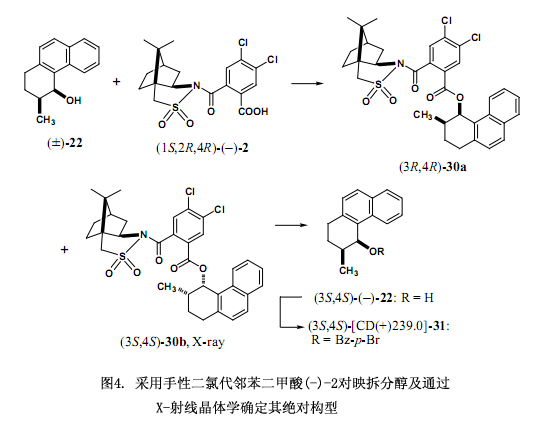
如图4所示的例子醇(±)-22在二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP)条件下用手性二氯代邻苯二甲酸(–)-2浓缩后获得两种酯的非对映体混合物,混合物再经过硅胶高效液相色谱[正己烷/乙酸乙酯=7:1,α = 1.18, Rs = 1.06]分离。酯30a在甲醇中重结晶后洗脱出的第一组分生成柔滑细微针形晶粒,不适于用X-射线分析,第二组分30b在乙醇中重结晶后生成较大的棱状晶体可用X-射线分析。采用2,10-樟脑磺内酰胺作内参比物以及用重原子效应都能清楚的确定30b的绝对构型是(3S,4S)型。LiAlH4还原酯30b来去掉手性辅助剂后生成对映纯醇(3S,4S)-(–)-2224,25),这种方法所测定的绝对构型与采用圆二色谱激子手性方法测定的结果相一致,这些方法也适用于测定相应的4 - 溴苯甲酸(31)的绝对构型24)。
近来我们还发现使用K2CO3/甲醇溶剂分解可以有效地从手性二氯代邻苯二甲酸酯获得对映纯醇,并具有较高的产率。
表3列举了其他一些应用实例,即使对位取代物决定着远离手性中心(如羧基上的碳原子)分子的手性,但是手性二氯代邻苯二甲酸酯类、对位取代二苯甲醇类25,26与34-38仍可以被硅胶高效液相色谱有效的分离29,30,32)。这些结果表明手性二氯代邻苯二甲酸很容易识别分子的手性,例如氢原子与对位取代官能团的差别。
手性识别(4-甲基苯基)-苯甲醇(36)中的醇官能团对于手性二氯代邻苯二甲酸酯就是不大可能的,因为手性分子中氢原子与甲基的差别不大,所以不能识别其手性。因而也无法利用硅胶高效液相色谱来分离非对映体。需要采用以下步骤来解决该问题,第一步:选择(4-溴苯基)(4'-甲苯基)甲醇(37)作为前驱体,用手性二氯代邻苯二甲酸对其进行对映分解,然后利用X-射线晶体学测定他们的绝对构型。把溴原子还原生成所要的对映纯醇(S)-(–)-3629),这一步是合成的关键,同位素取代的手性二苯甲醇的绝对构型的测定在下文叙述。
同位素取代物也可以产生分子手性,如2H取代二苯甲醇39上的氢原子,13C取代二苯甲醇43上的碳原子32,33)。不能直接用普通手性固定相高效液相色谱或者带有手性助剂硅胶高效液相色谱来对映分解这些同位素取代的手性化合物。诸如这个例子,首先必须选择一种前驱体如(±)-(4-溴苯基)(苯基-2,3,4,5,6-d5)甲醇(40),然后对映拆分二氯邻苯二甲酸并同时测定其绝对构型。既而将溴原子还原生成所需要的对映纯(苯基-2,3,4,5,6-d5)苯甲醇(39)32)。

利用以下步骤制备(苯基-2,3,4,5,6-d5)苯甲醇(39),采用手性二氯代邻苯二甲酸跟外消旋醇(±)-40缩合反应,随后采用硅胶高效液相色谱分离所生成的酯。经过一系列的重结晶过程两种非对映体酯都能生成针形晶体,先从第一组分41a回收对映纯醇(–)-40,另外还有部分(–)-40经樟脑酸性氯化物处理后转变成酯42。酯42具有良好的结晶度,生成适于X-射线分析的柱状晶体,采用(–)-樟脑酸的绝对构型作为内参比物测定醇部分的绝对构型为S型。然后在Pd-C存在的条件下利用H2NNH2/H2O还原醇(S)-(–)-40生成同位素取代的对映纯(苯基-2,3,4,5,6-d5)苯甲醇[CD(–)270.4]-(S)-39,并且可以明确的测定其绝对构型。利用波长为589nm的钠D线测算比旋光度[α]D来区分对映体,但是,很难测定同位素取代手性化合物的比旋光度[α]D。因此,我们提供一种采用圆二色谱数据测定对映体的新定义方法,由于圆二色谱不仅仅比比旋光度[α]D灵敏,而且在测定微量样品的时候更为精确。例如,[圆二色(–)270.4]-(S)-39在270.4nm处表明为负的圆二色对映体,测定其绝对构型为S型32)。
如果在一开始就使用(–)-樟脑酸作为对映拆分的手性助剂,那么这将变得更为有利,但是,(–)-樟脑酸的对映拆分能力比较弱。例如,由醇40生成的樟脑酸酯并不可以用硅胶高效液相色谱分离,因而有时依据不同的形势有必要选择两种手性助剂。
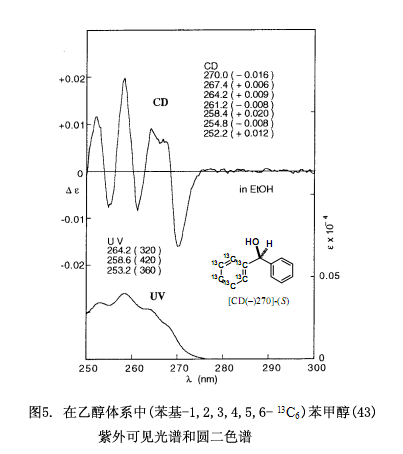
采用相似的合成步骤来合成13C同位素取代的手性二苯甲醇(43)33),也就是说,先选择(4-溴苯基)(苯基-1,2,3,4,5,6 - 13C6)甲醇(±)-(44),随后将其对映拆分并采用与上述描述相似的方式来测定它的绝对构型。再将溴原子还原生成13C取代的手性(苯基-1,2,3,4,5,6-13C6)苯甲醇[圆二色(–)270]-(S)-43。尽管由于同位素12C和13C之间有细微的差别而致的非常弱的分子手性,但是却能很容易的观察到13C取代的手性二苯甲醇(S)-43的圆二色谱,圆二色谱图如图5所示33)。
对于邻取代的二苯甲醇我们也获得了非常有趣的结果,使用以上描述的方法制备了对映体(2 -甲氧基苯基)苯甲醇(-)-(45),并测定其绝对构型为S型34)。以前采用不对称催化反应合成了手性(2-甲苯基)苯甲醇(46),基于手性反应机理估计了其绝对构型。但是,基于手性机理来确定的绝对构型结果是否可靠?在这样的疑问下就很有必要独立的、明确的来测定绝对构型。为了解决这个问题我们进行了以下实验,但不能成功的直接将46对映拆分成手性二氯代邻苯二甲酸,即使是有邻取代醇36存在也不行,因为很难区分邻位上的氢原子和甲基。然后我们尝试对映拆分(4-溴苯基(2’-甲苯基)甲醇(47),可是采用高效液相色谱分析时仅仅出现了一个峰。
然后我们采用以下间接方法34-36),此方法包括把(2-羟基-甲苯基)苯甲醇(48)对映拆分成手性二氯代邻苯二甲酸,用X-射线分析测定其绝对构型,将48的对映纯衍生物转化成所要的醇46。手性二氯代苯甲酸(–)-2(方程式1)可以与二醇(±)-48反应生成酯非对映体混合物,这些酯主要是通过对伯醇官能团进行的选择性酯化反应而合成的。在此情况下,手性助剂官能团与伯醇部分成键结合后距48的手性中心较远,因而很容易分离非对应酯。从第二部分(–)-49b可以获得单晶,用X-射线分析测定其绝对构型为S型。第一洗脱部分(R)-(–)-49a经过几步反应转变成所要的对映纯醇(R)-(–)-46,该结果显示先前采用基于不对称反应机理所确定46的绝对构型是错误的35),因为基于反应机理所确定的绝对构型有时可能产生错误,最明智的做法是分别测定产物的绝对构型。
这种方法还能有效的制备各种苄醇52-5537),这类醇还作为天然产物全部合成的手性合成子,原因是这些醇具有明确的绝对构型以及100%对映体纯度。
阿托异构体24是一个特殊的手性化合物,因为它含有三个萘发色团,其对映拆分和绝对构型的测定按下述方法执行27,28)。外消旋二醇、(±)-1,1':4',1'' 三萘-2,2''-二甲醇(24)加成到手性二氯代苯甲酸上形成一种酯类混合物,然后用硅胶高效液相色谱(正己烷/乙酸乙酯= 2:1, α = 1.18)分离。然而第一组分(–)-56a经正己烷/乙酸乙酯重结晶生成细微针形晶体,第二组分(+)-56b生成较大晶体。
总的来说,适用于X-射线分析的单晶必须是具有一定表面和边缘的棱状或者柱状晶体,也可以是厚板形式的晶体。酯(+)-56b经重结晶后生成具有跟飞机翅膀相似的三角形翅膀的晶体,这些晶体看起来不像单晶,将翅膀移开后剩余部分就是单晶,可以用X-射线测定主体部分的构型。更加有趣的是,由初步晶格常数估算的不对称单元的分子量跟(+)-56b分子量不一致,因此我们初步认为此分子的结构可能不正确,由手性二氯代邻苯二甲酸的不对称性部分可知,我们可以假设56b的不对称部分含有一个分子。认真考察所获得的数据可以看出56b分子的一半等价于一个不对称单元,也即是在酯(+)-56b晶体中有一个C2对称结构,尽管事实上此分子中含有复杂的手性二氯代邻苯二甲酸部分。例如利用内参比法测定扭转三萘生色团的绝对构型为(aS,aS),从酯(aS,aS)-(+)-56b上移去手性助剂生成对映纯二醇(aS,aS)-(+)-24,利用X-射线分析测定的绝对构型与采用圆二色谱激子手性方法测定的(+)-24绝对构型相符27,28)。

2-(1-萘基)丙基-1,2-二醇化合物作为在家兔体内1-异丙基萘的手性代谢产物而被分离,该代谢产物不是对映体,依据反应机理仅能凭经验估计其绝对构型。为了获得对映纯二醇57以及用一种清晰的方法测定其绝对构型,手性二氯代邻苯二甲酸方法可以用于(±)-57绝对构型的测定38)。在这种情况下,只有伯醇部分被酯化后得到非对映体混合物,再用硅胶高效液相色谱(正己烷/乙酸乙酯 = 4:1, α = 1.3, Rs = 1.1.)分离此混合物。在此高效液相色谱中自由叔醇官能团的存在尤为重要,但是对叔醇官能团的保护致使分离更加困难。
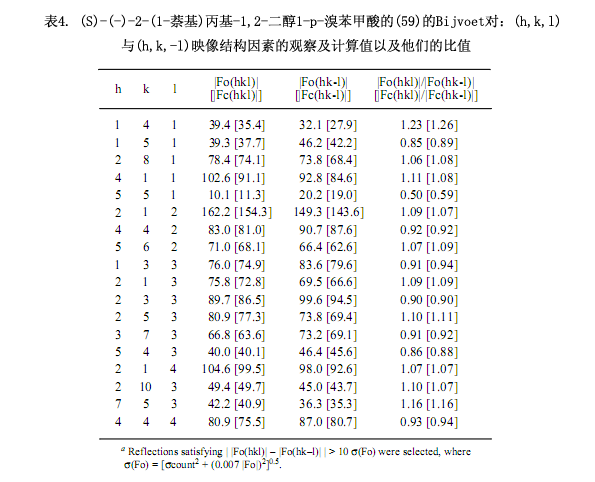
即使不断地重结晶,但是所得到的两种非对映体仅是无定形固体。用LiAlH4还原第一部分(–)-58a生成对映纯乙二醇(–)-57,然后进一步转化成4 - 溴苯甲酸(–)-59(图6.)。由乙醇对其重结晶,(–)-59能生成适合于X-射线分析的单晶,通过对含有溴原子的反常色散效应的Bijvoet对测量可以确切的确定其绝对构型为S型(表4)。
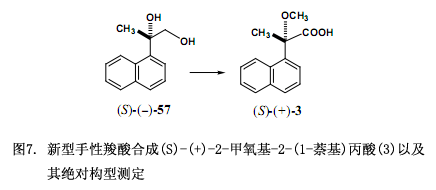
进一步说,我们通过几个反应步骤从二醇(S)-(–)-57获得对映体(S)-(+)-2-甲氧基-2-(1-萘基)丙酸(3)(图7)38)。如下讨论我们发现对对映拆分和绝对构型测定非常有效的新型羧酸39-41)。我们针对手性邻苯二甲酸(1)与二氯代邻苯二甲酸(2)应用研究与该文献(J. Synth.Org. Chem. Jpn. (Yuki Gosei Kagaku Kyokaishi)报道的相一致42)。
7.一种新型手性羧酸2-甲氧基-2-(1-萘基)丙酸(MαNP 酸),对醇的对映拆分与采用氢质子核磁共振各向异性方法测定其绝对构型的重要作用:基本原理及应用
我们已经探讨手性邻苯二甲酸与二氯代邻苯二甲酸的设计和应用,二者对于对映体化合物的合成以及采用X-射线分析明确的确定其绝对构型有着重要作用。然而表2和表3所列举的大部分应用仅限于芳香族化合物绝对构型的测定,是否有一种功能较强的方法来用于测定脂肪族化合物的绝对构型?
我们近来还发现新型手性羧酸2-甲氧基-2-(1-萘基)丙酸(MαNP酸 (3)),能有效的对映拆分脂肪醇,尤其是无环脂肪醇。由MαNP 酸3跟醇类反应生成的酯的氢质子核磁共振谱图可知,醇组分中的质子化学位移受萘官能团诱导的磁各向异性效应影响强烈38-41)。因而,此羧酸3在改进的Mosher方法中可以被用作手性助剂10),羧酸3对测定仲醇的绝对构型也非常有用。该羧酸3的另外一个优点就是它不具外消旋作用,因为3的α位是饱和的,以至于对映纯3很容易制备。以下讨论,MαNP 酸3是功能强的手性衍生剂,可以确保仲醇的对映拆分及其绝对构型的测定同时进行。这里说的MαNP 酸3在测定天然产物和合成具有生物活性的手性化合物(例如手性药物)的绝对构型时作用较强。从这种意义上来看,手性MαNP 酸3优于传统的手性酸Mosher’s MTPA酸10),Trost’s MPA酸12)以及由Riguera11)and Kusumi10)团队研发的1- and 2-NMA酸。
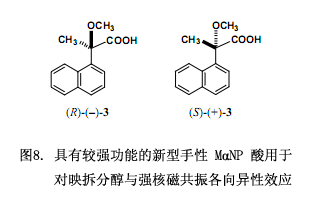
以下部分详细描述手性MαNP 酸3的基础理论和应用:手性酸3的合成;由X-射线与化学相关法测定其绝对构型;通过手性醇对映拆分外消旋酸3以及使用手性醇确定其绝对构型;采用核磁共振法与圆二色谱光谱法测定MαNP 酸酯的绝对构型和构象分析;用手性MαNP 酸3对醇对映拆分以及确定醇的绝对构型;从分离的非对映酯回收100%对映体纯度的手性醇;该方法应用于各种醇。
8.MαNP 酸3的简便合成,与天然(-)-薄荷醇结合后显著的对映拆分作用
为了合成大量对映纯手性8. MαNP 酸(3),图9显示了外消旋酸3的简便合成和对映拆分过程。总而言之,手性合成的胺或生物碱就是用于对映拆分羧酸,我们采用下面的新方法应用于手性醇,在此方法中用外消旋酸3来缩合手性醇,形成的非对应酯采用硅胶高效液相色谱分离,分离后的酯再经水解生成对映体和目标羧酸。
自然状态的薄荷醇作为一种手性醇而被选择,用外消旋酸3酯化该醇,我们发现很容易生成非对映酯63a与63b,再经硅胶高效液相色谱(正己烷/乙酸乙酯 =10:1)分离,其过程如图10所示。分离因数与分离度都很大(α= 1.83, Rs = 4.55),这同时还表明酸3有很强的识别醇手性的能力。酯63a先洗脱出来受到溶剂分解作用生成手性酸(+)-3,而63b随后被洗脱出生成酸(–)-3。为了测定所获得手性酸3的绝对构型先将其转化成甲酯,再用圆二色谱测定。把这些圆二色谱图跟X-射线分析与化学相关法测定的真实样品的绝对构型相对比,所得两种酯的绝对构型分别是(S)-(+) 和 (R)-(–),也就是(S)-(–)-63a和(R)-(–)-63b(图9所示)。

9.氢质子核磁共振各向异性方法测定仲醇的绝对构型:扇形规则及应用
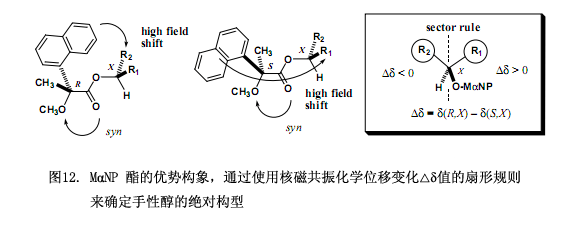
如上所述,氢质子核磁共振各向异性方法已经经常被用作一种相关的验证方法来测定手性有机化合物的绝对构型10-13),尤其是测定手性仲醇的改进Mosher方法已经成功用于测定天然产物的绝对构型。至于Mosher’s MTPA 酸与 Trost’s MPA 酸,其苯基呈现磁各向异性效应主要是因芳环环电流所致,再引起醇中质子的化学位移(δ)发生变化。因此,可以用(R)和(S)羧酸所形成的酯的化学位移变化(∆δ)(其中∆δ = δ(R) – δ(S) 或者 ∆δ = δ(S) – δ(R))来确定手性醇的绝对构型。我们发现MαNP 酸3要优于Mosher’s MTPA 酸和 Trost’s MPA 酸,原因是萘基的磁各向异性效应比苯基的磁各向异性效应要大,进而导致化学位移变化∆δ值增大,因此当采用MαNP 酸3作为手性核磁共振各向异性试剂,手性醇的绝对构型可以清楚的确定。
再者就是MαNP 酸还有另外一个优点即它本身不具有外消旋作用,因为它的α位上也是饱和的。从这些原因来看,在测定手性醇包括天然产物的绝对构型时,最好的做法就是使用MαNP 酸3而不是用传统的手性酸。
非对映体MαNP 酸酯63a和63b所有的核磁共振质子峰通过各种方法被分配开来,其中还包括二维方法(1H, 1H-1H COSY, 13C, 1H-13C COSY, HMBC, 图 11a)。酯63b中异丙基上的质子跟63a中的质子相比主要出现在高场区,另一方面63a中2位上的质子跟63b上的质子相比也出现在高场区。这些化学位移的明显变化主要是由于MαNP 酸中萘基的磁各向异性效应所致。

从核磁共振各向异性效应来确定绝对构型,需要确定每一个非对映体的优势构象。在酯63a和63b中MαNP 酸与薄荷醇的绝对构型已由上述方法确立,下面的稳定构型主要是满足从核磁共振图谱中观察到的各向异性效应(图11)。也即是在它们各自稳定的构型中,甲氧基跟酯羰基里面的两个氧原子相互发生顺叠,还有酯羰基氧原子顺叠到醇甲烷质子上,至此甲氧基、酯基以及醇甲烷质子在同一平面上,该平面被称作MαNP平面(图11)。这些顺叠构象跟MPA酯里面的构象相似,在酯63a中萘基和H-2质子都位于MαNP平面的同一前侧,并且H-2质子位于萘基之上,因而H-2质子受到高场位移的磁各向异性效应,所以出现在高场区;在酯63b中萘基与异丙基相距较近,可以观察到异丙基质子具有高场位移。
酯63a和63b顺叠构象的优势还可以通过对比其核磁共振数据跟2-羟基-2-(1-萘基)丙酸薄荷酯的核磁共振数据(图11(b)所示)来看出。从核磁共振化学位移及红外数据可以很明显的看出叔醇羟基官能团中的氢原子键合到酯羰基的氧原子上,也即是羟基与酯羰基氧原子产生顺叠构象,我们还发现一个有趣的现象2-羟基-2-(1-萘基)丙酸薄荷酯(S;1R,3R,4S)-(–)-63a的核磁共振化学位移数据尤其是薄荷醇部分的数据跟(图11(a)、(b)所示)2-羟基-2-(1-萘基)丙酸薄荷酯(S;1R,3R,4S)-(–)数据极其相似。对其他非对映体对来说亦是如此,例如(R;1R,3R,4S)-(–)-63b2-羟基-2-(1-萘基)丙酸薄荷酯(R;1R,3R,4S)-(–)也是这样的结果。这些事实进一步说明MαNP酸薄荷酯呈现顺叠构象,HαNP酸薄荷酯也是这样,而且这都完全可以用磁各向异性效应来解释。
根据采用MαNP酯核磁共振各向异性效应,我们可以引出适用于确定仲醇绝对构型的扇形规则(图12)。基本步骤如下:(R)-MαNP酸 和(S)-MαNP酸分别与一种手性醇反应,将其绝对构型定义为X型,因此由(R)-MαNP酸所制得酯具有(R,X)绝对构型,另外一个由(S)-MαNP酸制得酯绝对构型为(S,X)型。(R,X)- 酯和(S,X)-酯所有的核磁共振质子信号经仔细分析后被完全分配,如有必要我们建议使用二维光谱。化学位移变化∆δ值((∆δ= δ(R,X) – δ(S,X))适用于计算醇中的所有质子。图12显示了用于MαNP酯的扇形规则,在这里面MαNP基团放置在前下方,而仲醇中甲烷质子放置在后下方;带有化学位移变化∆δ正值的R1基团质子放置在右边,而带有化学位移变化∆δ负值的R2基团质子放置在左边。从这个投影上看,也就可以确定手性醇的绝对构型是X型。
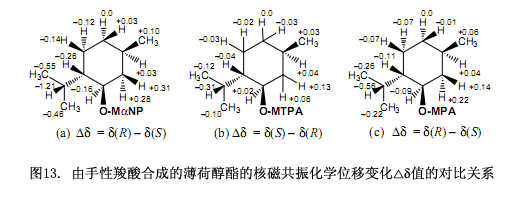
手性MαNP酸的磁各向异性效应比常用手性羧酸的磁各向异性效应强(图13),例如MαNP酸薄荷酯的化学位移变化∆δ值大约是Mosher’s MTPA酯的∆δ值的四倍还要大10)(图13b),大约是Trost’s MPA酯的两倍12)(图13c),是Riguera11)和 Kusumi10)等人报道的2-NMA酸酯的∆δ值的两倍。因此,MαNP酸酯可以有效的测定天然产物的绝对构型。
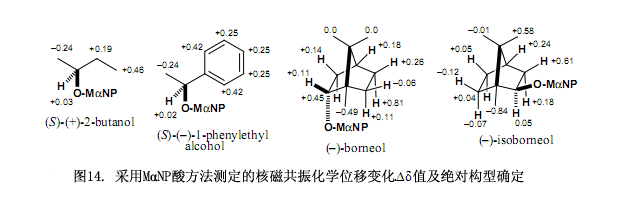
此MαNP酸方法在其他手性醇上的应用如图14所示。
10.使用MαNP酸对不同醇的手性拆分及其绝对构型的同时测定41)
MαNP酸的另外一个优点就是其具有较强的手性识别能力,例如正如以上所讨论的外消旋MαNP酸可以成功的对映拆分天然(–)-薄荷醇酯。所生成的非对映酯能够采用硅胶高效液相色谱清楚的分离,MαNP酸可以对映拆分如图14所列出的其他手性醇,这些事实有逻辑地表明如果使用对映纯MαNP酸可以对映拆分外消旋醇,事实上我们成功的运用(S)-(+)-3MαNP酸对映拆分了不同醇,例如图15所示。

新型手性(S)-(+)-3 MαNP酸具有功能显著对映拆分醇的能力,尤其是对映拆分手性醇。例如2-丁醇所合成的非对映酯在分离因数α = 1.15,分离度Rs = 1.18时基本上都能被分离,在此情况下手性羧酸3就可以明显的识别仅有细微差别的甲基和乙基官能团。因为手性酸3对脂肪醇呈现出很强的分辨能力所以该方法是一种优越而又实用的方法,还可以用于几乎不可能实现的的不对称合成反应上。
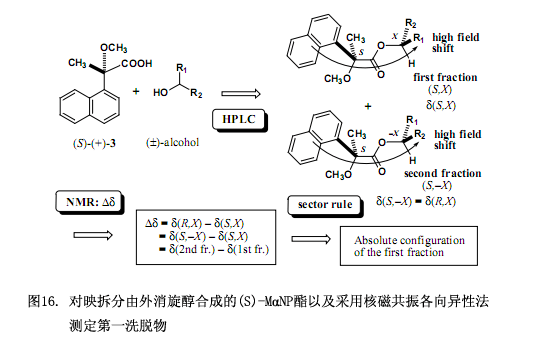
而后另外的问题是怎样测定醇部分的绝对构型,用上述描述的手性MαNP酸依据核磁共振各向异性效应测定所分离的非对映体的绝对构型。其总体方案如图16所示,用(S)-(+)-3MαNP 酸酯化外消旋醇生成非对映酯混合物,再用硅胶高效液相色谱分离此混合物。第一洗脱物的绝对构型被定为(S,X),这里的S表示MαNP酸部分的绝对构型,而X表示醇部分的绝对构型。酯第二洗脱物的绝对构型表示为(S,–X),此处的-X为X相反的绝对构型。化学位移变化∆δ值的最初定义是∆δ = δ(R,X) – δ(S,X),若要计算∆δ值必须先知道δ(R,X)的值,然而此方案中不存在对映体(R,X),因此化学位移变化∆δ的原始方程在这里没有多大用处。
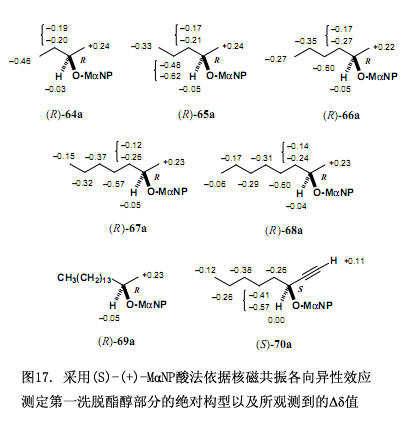
为了解决以上问题需要对方称进行如下改变,因为酯(S,–X)是酯(R,X)的对映体,因而他们的核磁共振数据彼此相同,也即∆δ = δ(R,X) – δ(S,X)= δ(S,–X) – δ(S,X) = δ(2nd fr.) – δ(1st fr.),第一洗脱部分的绝对构型X可以由化学位移变化∆δ值来确定,此∆δ值也就是用第一洗脱部分的化学位移值来减去第二洗脱部分的化学位移值而得到(图16)。本方法已经用于图15所示的酯,可以得到∆δ值及第一洗脱酯的绝对构型(图17),这些∆δ值已被合理分布,右边是正值,左边是负值,因而可以确定第一洗脱酯的绝对构型,相反的绝对构型就是第二洗脱酯的了。所需要注意的是如果采用(R)-(–)-3MαNP 酸,那么就应该这样定义∆δ值,∆δ = δ(R,X) – δ(S,X)= δ(R,X) – δ(R,–X) = δ(1st fr.) – δ(2nd fr.) 。
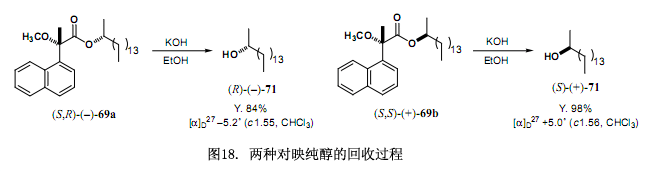
第二步是回收对映纯醇与手性MαNP 酸3如图18 例子所证实,分离后的酯经溶剂分解作用生成对映纯醇。回收的手性MαNP酸3还可以重复利用。
什么是回收醇的对映体纯度?在我们看来所获得两种非对映酯都是对映纯的,因为如果我们使用的MαNP酸3是对映纯的,他们能很好的用高效液相色谱分离开。MαNP酸3能够用天然(–)-薄荷醇对映拆分,用手性固定相的气相色谱测定,其对映体纯度肯定是100%。
如上所述,即使不考虑分子结构的简单性,MαNP酸也具有优越的对映拆分能力,在磁各向异性效应方面手性酸3要优于Mosher’s MTPA 酸与Trost’s MPA 酸,因而需要进一步的讨论。
11.通过氢质子核磁共振或者质谱法的新非对应异构体法来测定对映体过量43)
怎样通过不对称合成或者酶促反应确定所获得的手性化合物的对映体过量(% ee)?因为手性分子化学在有机合成中的重要性持续增加,因而急需要新型的、准确的方法来测定对映体过量。对映体过量(% ee)有如下定义:
% ee = 100{ | (R) – (S) | }/{(R) + (S)} (1)
上面已经说到我们开发了一种新的手性磁各向异性试剂MαNP 酸3(图19),它是一种通过氢质子核磁共振谱图来测定手性醇绝对构型的较强功能试剂38-41)。手性酸3的另一个特点就是它具有很强的对映拆分外消旋醇的能力,特别是通过高效液相色谱对映分解无环脂肪醇,诸如MαNP酯类。例如经MαNP酸 (S)-(+)-3酯化得到的消旋2-十六醇(±)-71,用硅胶高效液相色谱(正己烷/乙酸乙酯 = 20:1;分离因数α = 1.93 和分离度 Rs = 3.68)很容易分离所获得的非对映体混合物。经过酯的溶剂分解作用很容易得到2-十六醇-71的两个纯的对映体,这两个对映体分别作为第一、第二洗脱组分被洗脱出来。MαNP 酸3的这些优良特性有助于我们利用氢质子核磁共振和质谱图法来开发新的非对映体法测定对映体过量43)。

有很多种测定手性化合物对映体过量的方法:1)对比对映体化合物的比旋光度[α]D值或圆二色谱强度;2)采用手性固定相的高效液相色谱或气相色谱进行对映分离44);3)利用手性有机金属位移试剂的氢质子核磁共振检测45);4)通过高效液相色谱或氢质子核磁共振检测来分离由手性衍生剂所合成的非对映体46);5)通过利用手性衍生剂或手性主-客体络合物做质谱检测47-50);使用对映分解动力学方案的方法51)。以上这些方法都具有各自的优缺点,例如在一些情况下通过使用标准样品的已知对映体过量值来确定对映体化合物的数据和校准曲线;在另外一些情况下峰变宽就扰乱了峰值强度的精确测定。
对于使用手性衍生剂的非对映体方法,总会伴随着动力学拆分的影响,因此最重要的问题是如何评价这种方法。如果衍生化反应获得了100%产率,那么就可以排除动力学拆分影响;但是,反应要达到100%的产率是不切实际的。另外,基于动力学分解效应的方法包括近似方法和需要校准曲线;在个别情况下依据以下粗略近似来测定对映体过量。手性衍生剂的两个对映体分别与手性底物反应,我们检测了所生成的产物的产率,当它们各自反应的产率相似时,可以简单地认为动力学拆分影响可以忽略不计,不用对动力学拆分效应进行校正就能直接计算对映体过量的值。用非对映方法测定对映体过量时经常受动力学拆分效应干扰,不过就像下面所说,在彻底消除动力学拆分效应的情况下我们依据氢质子核磁共振与质谱已经成功的率先发展一种新型的对映体法来测定对映体过量43)。我们研发的方法优点之一是使用质谱保证了微量成分的高灵敏度检测。
12.测定对映体过量新型方法的原则和步骤:彻底消除动力学拆分效应
利用2-十六醇71作为试验样品来解释测定对映体过量新型方法的原则和步骤,如图20所示一份大约1:1的(S)-(+)-3MαNP 酸和氘标记的(R)-(–)-3-d3 MαNP酸混合物与手性醇71反应(0-100%的对映体过量)生成酯的非对映体混合物(图 20, R = H, n = 3)。
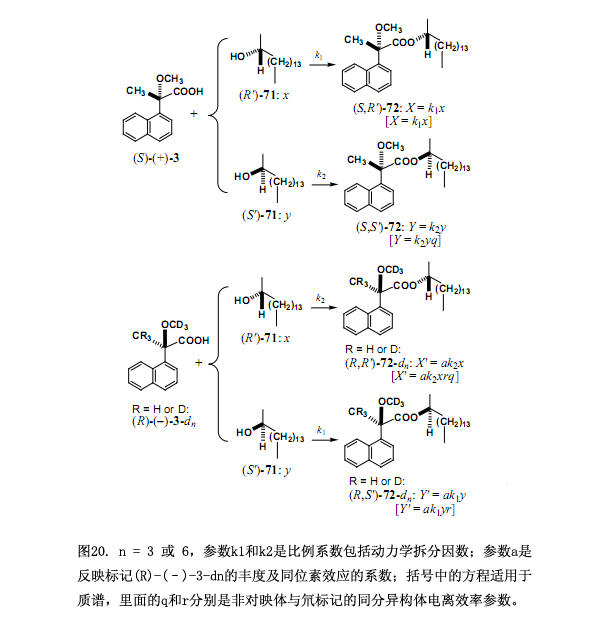
(R')-71 和(S')-71的组成分别定为x和y,其中x + y = 1,同样的所生成的酯的组分被定为:酯 (S,R')-72, X;(S,S')-72, Y; (R,R')-72-d3, X';(R,S')-72-d3, Y'。酯的总量用以下公式表示:X = k1x 和 Y = k2y,在这里k1、k2是比例系数包括动力学拆分因数(注意这些系数都不是速率常数),由于酯(S,S')-72 和(R,R')-72-d3相互是对映体,相同的系数k2用来定义(R,R')-72-d3的量:X' = ak2x,此处a是反映形成酯的(R)-(–)-3-d3丰度与同位素效应的系数;对于另一个酯(R,S')-72-d3, Y' = ak1y。采用比例X/Y',这些方程简化成方程(1),方程(1)中消去了比例系数也包括动力学拆分因数的k1。
X/Y' = (k1x)/(ak1y) = (1/a)x/y (2)
同样的k2也被消去了
X'/Y = (ak2x)/(k2y) = (a)x/y (3)
由方程(2)与方程(3)作用得到方程(4),这里反映(R)-(–)-3-d3丰度和同位素效应的系数a也被消去了。结果等于醇组分比(x/y)的平方。
(X/Y')(X'/Y) = [(1/a)x/y][(a)x/y] = (x/y)2
= [(1st,M)/(1st,M+3)][(2nd,M+3)/(2nd,M)] (4)
利用硅胶高效液相色谱(22φ × 300 mm,正己烷/乙酸乙酯 = 20:1)分离这些非对映体酯的混合物(图21)。第一部分包含酯(S,R')-72 和(R,S')-72-d3,可以从MαNP部分的甲氧基与甲基官能团的氢质子核磁共振峰强度来确定各组分比例(X/Y') = [(1st,M)/(1st,M+3)],也即是甲氧基的峰强度对应X,而甲基的峰强度对应X+Y';相似的处理也适用于含有(S,S')-72 和(R,R')-72-d3的组分2,由氢质子核磁共振峰强度得到比值(X'/Y) = [(2nd,M+3)/(2nd,M)],将上面的比值带入到方程(4)利用x+y = 1的关系中计算(x/y)2,因而就确定了醇71的对映体过量(% ee)。
13.氢质子核磁共振测定对映体过量应用(% ee)
甲基-2-羟基-2-(1-萘基)丙酸跟CD3I(氘原子组分>99.5 %)经甲基化反应生成氘标记的(R)-(–)-3-d3MαNP 酸,随后水解并经(–)-薄荷醇对映拆分。(S)-(+)-3 和(R)-(–)-3-d3混合物(比, 1:0.987, 总量8.1 mg, 0.0349 mmol)可以与测试样品手性2 -十六醇(71, 9.237 mg, 1.09 × 0.0349 mmol, 60.9% 对映体过量按重量计算)反应,生成酯72的非对映体混合物,该混合物再由硅胶高效液相色谱(正己烷/乙酸乙酯 20:1)分离(图21)。
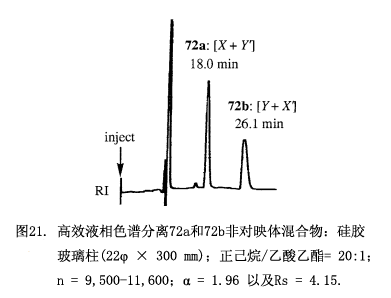
从第一组分的氢质子核磁共振图谱可以确定酯(S,R')-72和 (R,S')-72-d3的组成:X = 1.00, Y' = 0.22, X/Y' = 4.54,类似的从第二组分的氢质子核磁共振图谱确定(S,S')-72 和 (R,R')-72-d3 的组成:Y = 1.00, X' = 3.60, X'/Y = 3.60。结果为(X/Y')(X'/Y) = 16.3636,x/y = 4.045,再根据x+y = 1得出x = 0.8017 及 y = 0.1982,并获得60.4%对映体过量,这个值与按重量计算的对映体过量60.9%相符合,本方法已经用于10种71测试样品,其对映体过量从0-90%。如图22所示由氢质子核磁共振所获得的对映体过量与按重量计算的值相一致,平均误差为±0.4% 对映体过量,而最大误差1.3% 对映体过量。这些结果清楚的验证了我们测定对映体过量方法的方案和原理。当我们将5种测试样品的对映体过量扩大到92-100%时,其偏差变大:平均误差±1.2% 对映体过量,最大误差6.3%对映体过量。如此大的偏差可能是由于这些检测仅限于测试氢质子核磁共振弱峰的强度。为了克服这个问题我们采用更为灵敏的质谱来测定组成。

14.质谱确定对映体过量及应用
就质谱而言,非对映体(S,R')-72和(S,S')-72的电离效率不尽相同,氘取代酯(R,S')-72-dn与非取代酯(S,R')-72也不同。因此各异构体电离效率因数(f)定义如下:酯(S,R')-72,f = 1,X = k1x;酯(S,S')-72,f = q 以及Y = k2yq;(R,S')-72-dn (n = 3 或 6), f = r 与Y' = ak1yr;(R,R')-72-dn (n = 3或6), f = rq 与 X' = ak2xrq。在这里由于非对映体结构的差别q是相对电离效率,r跟氘取代有关;做比值X/Y',方程(1)就变成:
X/Y' = (k1x)/(ak1yr) = (1/ar)x/y (2')
类似的方程(2)变形为
X'/Y = (ak2xrq)/(k2yq) = (ar)x/y (3')
因此,方程(3)变为
(X/Y')(X'/Y) = [(1/ar)x/y][(ar)x/y] = (x/y)2
= [(1st,M)/(1st,M+n)][(2nd,M+n)/(2nd,M)] (4')
由于所有的电离效率系数都被取消掉,测定对映体过量同样的方案适用于质谱测定。
采用质谱测定对映体过量的方法基本适用于三氘取代的(R)-(–)-3-d3MαNP酸,在质谱中3个m/z单位(M+3 vs. M)不足以避免自然丰度同位素峰的重叠,因此我们需要做一系列的修正,在修正背景后,跟同位素峰重叠,氘的含量,外消旋体(0%对映体过量),在(0-100% 对映体过量)范围内都很好的符合:平均误差为±0.7%对映体过量;最大误差是1.9%对映体过量。然而,这些修正针对实际应用就太复杂了,因此我们选择了六氘取代的(R)-(–)-3-d6MαNP 酸。
2-(1-萘基)-2-乙醛酸乙酯和CD3I (氘原子含量 >99.5 %)经Grignard反应制备了对映体手性酸(R)-(–)-3-d6,然后用CD3I (氘原子含量>99.5 %)对其甲基化反应,再经水解、用(–)-薄荷醇对映拆分。对于此手性酸(R)-(–)-3-d6,由于6个m/z单位(M+6 vs.M)的差别,自然丰度的同位素峰重叠完全可以忽略不计。

接下来使用测试样品71来检查这MS光谱法(按重量计算69.77%对映体过量),由(S)-(+)-3和(R)-(–)-3-d6(二者比:1:0.994)制备非对映体酯混合物,该混合物经硅胶高效液相色谱(正己烷/乙酸乙酯 20:1)分离。从第一组分的质谱图23(a)可以看出,用背景校正法测得酯(S,R')-72 (M+ m/z 454) 和酯 (R,S')-72-d6 (M+ m/z 460)的组成:X = 52.284, Y' = 11.221, X/Y' = 4.6594;类似的从第二组分的质谱图23(b)来看(S,S')-72 (M+ m/z 454)和(R,R')-72-d6 (M+ m/z 460)的组成:Y = 8.607,X' = 55.278, X'/Y = 6.4224。结果是(X/Y')(X'/Y) = 29.9245, x/y = 5.4703. 由于 x+y = 1,所以得出x = 0.845448及y = 0.154552,还得到69.09%对映体过量,此对映体过量跟用按重量法计算的对映体过量69.77%相吻合。
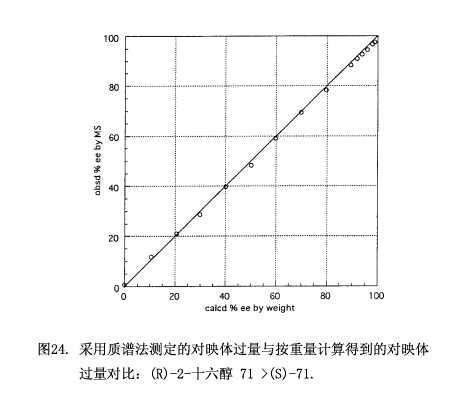
质谱法已经用于测定如图24所示的15种对映体过量0-100%的测试样品71,由质谱法所得到的对映体过量值跟按重量计算的对映体过量值相一致,平均误差是±1.08%对映体过量,最大误差为1.79%对映体过量;与氢质子核磁共振图谱不同的地方就是,即使在90-100%对映体过量范围内数据点也呈直线关系。质谱法要比氢质子核磁共振法灵敏,因此适用于测定高对映体过量的样品。尽管我们做的样品(R)-2-十六醇 71 > (S)-71,但是按同样的方案也可以得出样品(S)-2-71 > (R)-71。
因此,在彻底消除动力学拆分效应情况下我们成功的开发了新的非对映体法来测定对映体过量43),这种方法中值得强调的是其既不需要对映纯化合物数据也不需要制备使用已知对映体过量样品校准曲线,还不需要动力学拆分效应数据。该法的优越性就是以采用高效液相色谱分离非对映体为代价所得到。此方法还需在精确度上进一步改进,现在正在调查研究除了醇以外还能否用于其他的化合物,Chemistry and Biology (Kagaku to Seibutsu)52)文献已经报道了这部分的详细描述。
15.结论
我们研发了几种手性助剂,尤其是手性羧酸。利用非对映高效液相色谱法成功的运用这些手性衍生剂对映拆分了醇,通过X-射线晶体学或者氢质子核磁共振各向异性法测定了其绝对构型,根据氢质子核磁共振与质谱法测定了对映体过量。X-射线晶体学法使用一个内参比物是测定绝对构型的最佳方法,但是很难得到理想单晶。在这种情况下,使用MαNP酸的氢质子核磁共振法,该酸无需结晶过程就能的有效的测定绝对构型。在对映拆分中手性二氯代邻苯二甲酸与MαNP酸二者互补,当用一种手性衍生剂拆分失败后,就考虑用另外一个酸。上述方法在实验室制备100%对映体纯度对映纯醇功能甚强,并能同时测定其绝对构型。进一步说,我们的方法适用于各种化合物,对映体的大量合成以及现在所进行的提升质谱测定对映体过量的质量。
参考文献
1) Bijvoet, J. M.; Peerdeman, A. F.; Van Bommel, A. J. Nature 1951, 168, 271.
2) Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy-Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, Calif., and Oxford University Press: Oxford, 1983.
3) Harada, N.; Soutome, T.; Murai, S.; Uda, H. Tetrahedron: Asymmetry 1993, 4, 1755.
4) Harada, N.; Soutome, T.; Nehira, T.; Uda, H.; Oi, S.; Okamura, A.; Miyano, S. J. Am. Chem. Soc. 1993, 115, 7547 .
5) N.; Hattori, T.; Suzuki, T.; Okamura, A.; Ono, H.; Miyano, S.; Uda, H. Tetrahedron: Asymmetry 1993, 4, 1789.
6) Hattori, T.; Harada, N.; Oi, S.; Abe, H.; Miyano, S. Tetrahedron: Asymmetry 1995, 6, 1043.
7) Toda, F. Top. Curr. Chem. 1987, 140, 43. Toda, F. Inclusion Compounds; Atwood, J. L.; Davis, J. E.; MacNicol, D. D., Ed.; Oxford University Press: Oxford, 1991; Vol. 4, 126-187. Toda, F. Advances in Supramolecular Chemistry; Gokel, G. W., Ed.; JAI Press: London, 1992; Vol. 2, 141-191.
8) Toda, F.; Tanaka, K.;Miyahara, I.; Akutsu, S.; Hirotsu, K. J. Chem. Soc., Chem. Commun. 1994, 1795. Toda, F.; Tanaka, K.; Leung, C. W.; Meetsma, A.; Feringa, B. L. J. Chem. Soc., Chem. Commun. 1994, 2371. Toda, F.; Tanaka, K.; Watanabe, M.; Abe, T.; Harada, N. Tetrahedron: Asymmetry 1995, 6, 1495-1498.
9) Toda, F. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1990, 47, 1118.
10) Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092. Kusumi, T.; Takahashi, H.; Ping, X.; Fukushima, T.; Asakawa, Y.; Hashimoto, T.; Kan, Y.; Inouye, Y. Tetrahedron Lett. 1994, 35, 4397. Kusumi, T.; Takahashi, H.; Hashimoto, T.; Kan, Y.; Asakawa,Y. Chem. Lett. 1994, 1093.
11) Seco, J. M.; Latypov, Sh. K.; Quinoa, E.; Riguera, R. Tetrahedron Lett. 1994, 35, 2921. Latypov, Sh. K.; Seco, J. M.; Quinoa, E.; Riguera, R. J. Org. Chem. 1995, 60, 504. Seco, J. M.; Latypov, Sh. K.; Quinoa, E.; Riguera, R. Tetrahedron 1997, 53, 8541.
12) Trost, B. M.; Belletire, J. L.; Godleski, S.; McDougal, P. G.; Balkovec, J. M.; Baldwin, J. J.; Christy, M. E.; Ponticello, G. S.; Varga, S. L.; Springer, J. P. J. Org. Chem. 1986, 51, 2370.
13) Orui, H. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1998, 56, 591. Fukushi, K. Nippon Nogeikagaku Kaishi 1998, 72, 1345.
14) Review, J. Chromatogr. A 2001, 906, 1-482. Yamamoto, C.; Okamoto, Y. Maku 2000, 25, 277. Kobayashi, Y.; Matsuyama, A.; Onishi, A. Fine Chemicals 2000, 29, 59.
15) Stereoselective Biocatalysis; Patel, R. N. Ed.; Marcel Dekker: New York, 2000.
16) Harada, N.; Ochiai, N.; Takada, K.; Uda, H. J. Chem. Soc., Chem. Commun. 1977, 495.
17) Harada, N.; Ono, H.; Nishiwaki, T.; Uda, H. J. Chem. Soc., Chem. Commun. 1991, 1753.
18) Weismiller, M. C.; Towson, J. C.; Davis, F. A. Organic Syntheses 1990, 69, 154.
19) Nehira, T.; Harada, N. to be published.
20) Toyota, S.; Miyasaka, T.; Matsumoto, Y.; Matsuo, T.; Oki, M. Bull. Chem. Soc. Jpn. 1994, 67, 1680.
21) Toyota, S.; Akinaga, T.; Kojima, H.; Aki, M.; Oki, M. J. Am. Chem. Soc. 1996, 118, 11460.
22) Toyota, S. Enantiomer 1999, 4, 25.
23) Harada, N.; Nehira, T.; Soutome, T.; Hiyoshi, N.; Kido, F. Enantiomer 1996, 1, 35 .
24) Harada, N.; Koumura, N.; Robillard, M. Enantiomer 1997, 2, 303.
25) Harada, N.; Koumura, N.; Feringa, B. L. J. Am. Chem. Soc. 1997, 119, 7256.
26) Toyota, S.; Yasutomi, A.; Kojima, H.; Igarashi, Y.; Asakura, M.; Oki, M. Bull. Chem. Soc. Jpn. 1998, 71, 2715.
27) Harada, N.; Vassilev, V. P.; Hiyoshi, N. Enantiomer 1997, 2, 123.
28) Harada, N.; Hiyoshi, N.; Vassilev, V. P.; Hayashi, T. Chirality 1997, 9, 623.
29) Harada, N.; Fujita, K.; Watanabe, M. Enantiomer 1997, 2, 359.
30) Fujita, K.; Harada, N. to be published.
31) Koumura, N.; Harada, N. to be published.
32) Harada, N.; Fujita, K.; Watanabe, M. Enantiomer 1998, 3, 64.
33) Harada, N.; Fujita, K.; Watanabe, M. J. Phys. Org. Chem. 2000, 13, 422.
34) Kuwahara, S.; Watanabe, M.; Harada, N.; Koizumi, M.; Ohkuma, T. Enantiomer 2000, 5, 109.
35) Watanabe, M.; Kuwahara, S.; Harada, N.; Koizumi, M.; Ohkuma, T. Tetrahedron: Asymmetry 1999, 10, 2075.
36) Taji, H.; Harada, N. to be published.
37) Kosaka, M.; Watanabe, M.; Harada, N. Chirality 2000, 12, 362.
38) Kuwahara, S.; Fujita, K.; Watanabe, M.; Harada, N.; Ishida, T. Enantiomer 1999, 4, 141.
39) Ichikawa, A.; Hiradate, S.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N. Tetrahedron: Asymmetry 1999, 10, 4075.
40) Harada, N.; Watanabe, M.; Kuwahara, S.; Sugio, A.; Kasai, Y.; Ichikawa, A. Tetrahedron: Asymmetry 2000, 11, 1249.
41) Taji, H.; Kasai, Y.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N.; Ichikawa, A. Chirality 2002, 14, 81.
42) Harada, N.; Watanabe, M.; Kuwahara, S.; Kosaka, M. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 2001, 59, 985.
43) Taji, H.; Watanabe, M.; Harada, N.; Naoki, N.; Ueda, Y. Org. Lett. 2002, 4, 2699.
44) Okamoto, Y.; Hatano, K.; Aburatani, R.; Hatada, K. Chem. Lett. 1989, 715.
45) Kabuto, K.; Sasaki, Y. J. Chem. Soc., Chem. Commun. 1987, 670.
46) Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512.
47) Sawada, M.; Yamaoka, H.; Takai, Y.; Kawai, Y.; Yamada, H.; Azuma, T.; Fujioka, T.; Tanaka, T. Int. J. Mass Spectrom. 1999, 193, 123. Sawada, M.; Takai, Y.; Iwamura, H.; Yamada, H.; Takahashi, S.; Yamaoka, H.; Hirose, K.; Tobe, Y.; Tanaka, J. Eur. J. Mass Spectrom. 2001, 7, 447.
48) Guo, J.; Wu, J.; Siuzdak, G.; Finn, M. G. Angew. Chem. Int. Ed. 1999, 38, 1755 .
49) Grigorean, G.; Ramirez, J.; Ahn, R. H.; Lebrilla, C. B. Anal. Chem. 2000, 72, 4275.
50) Yao, Z.-P.; Wan, T. S.; Kwong, K.-P.; Che, C.-T. Anal. C
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号