有机修饰及有机物对硅氧烷溶胶-凝胶过程的影响
- 2012-06-11
- 专题
早在 18 世纪中期,人们利用小分子量的四乙氧基硅烷(TEOS)和四甲氧基硅烷(TMOS) 作为前驱体经由溶胶-凝胶过程制备质地均匀的玻璃、陶瓷等材料[1]。目前,经由溶胶-凝胶过程制备的有机-无机复合材料广泛地应用于光学器件、电子元件、化学传感、信息存储、功能涂层和薄膜等领域[2]。为了更好地理解和控制材料的制备条件,人们借助各种表征手段如核磁共振(1H NMR[3]、13C NMR[4]、29Si NMR[5])、FTIR[6]、Raman[7]、SAXRD 和WA X R D[8]、AFM[9]、凝胶渗透色谱[10]、TEM和SEM[11]、光物理手段[12]等对于溶胶-凝胶过程进行大量的机理研究。溶胶-凝胶过程包含水解反应和缩聚反应两种基本化学反应,硅氧烷水解过程为 SN2 机理[13,14],由此可知取代基的性质及结构必然对水解和缩合反应产生影响;此外,引入的有机组分如能与水解或缩合产物发生相互作用,也必然会影响前驱体的溶胶-凝胶过程。为了较系统地介绍这方面的最新研究动态,本文重点综述关于有机修饰及有机物对硅氧烷前驱体溶胶-凝胶过程影响以及改性机制的研究。
1 有机修饰硅氧烷前驱体对溶胶-凝胶过程影响
内源性有机修饰硅氧烷是指在小分子硅氧烷前驱体如 TEOS 或TMOS 的基础上,有机基团取代烷氧基生成不可水解的 Si-C键。不同于小分子前驱体,经过有机修饰的硅氧烷因为取代基结构和性质的差异会导致其水解-缩聚反应速度、产物分布、空间结构演化、组织排布方式、产物存在状态、产物微区环境等方面的差异。
Piana 等[10]利用凝胶渗透色谱跟踪3-缩水甘油醚丙基三甲氧基硅烷(GPTMS)和甲基丙烯酰氧丙基三甲氧基硅烷(MAPTMS)在相同条件下水解产物分子量的分布,结果发现两种前驱体水解-缩聚反应产物的物种(单体、低聚物、高聚物)分布各不相同。Fyfe 等[15]利用高分辨率29Si NMR 对甲基三乙氧基硅烷(MTES)和小分子前驱体四乙氧基硅烷(TEOS)混合体系溶胶-凝胶过程的初始阶段进行了动力学分析。结果发现,MTES 单体烷氧基每一步水解速度比 TEOS 中烷氧基每一步水解速度快,他们认为甲基的存在对水解产物过渡态具有稳定作用。为了探讨有机取代基对硅氧烷溶胶-凝胶过程的影响,Hook[5]对多种有机取代硅氧烷的水解-缩聚速度进行了系统研究,利用诱导效应及立体效应机制解释取代基对水解-缩聚速度的影响。硅原子的d 轨道和氧原子之间可形成反馈键,硅原子上的取代基影响其 d 电子向氧原子反馈,必将影响到氧原子的碱性,即质子化程度。相对乙氧基而言,甲基具有给电子效应,增加硅原子上的电负性,继而增强氧原子的碱性,提高质子化程度。因此导致甲基取代硅氧烷水解速度较快。相对甲基而言,乙烯基较弱的诱导效应降低了烷氧基的质子化程度,从而使反应速度减慢。由于前驱体水解过程为 SN2 机理,不难理解苯基较大的位阻效应会导致反应前驱体活性降低。
取代基不仅影响水解-缩聚反应的动力学过程,而且这种影响还可导致反应溶胶-凝胶产物的空间结构衍化表现各异。Sugahara 等[16]研究了Si(OEt)4-RSi(OEt)3-EtOH-Water-HCl(R=Me, Ph) 体系在水解-缩聚的起始阶段 R 基团对缩合反应生成共聚体的影响。在富水体系中,含有较为疏水的苯基的水解产物在溶液中易于自簇集,而含有甲基前驱体的水解产物易于分散,从而导致甲基三甲氧基硅烷和四甲氧基硅烷水解产物可以形成共聚体,而苯基三甲氧基硅烷和四甲氧基硅烷的水解产物之间不能共聚,只能各自进行缩合反应。说明在利用不同取代基硅氧烷制备共聚杂化材料时,必须依据取代基疏水性选择水量的多少,以保证不同取代基硅氧烷水解产物的共聚。Matĕjka 等[8]研究了包括高聚物取代基在内的有机取代基对硅氧烷前驱体溶胶-凝胶过程的影响,认为聚合物的生长受控于分子之间缩聚反应和环化的竞争,有机取代基的链越长,越容易形成多面体的环形低聚物,主要是T8环或更大的笼状结构产物。因此较长的取代基增加了低聚物的稳定性,阻碍了缩聚反应,因此体系难以形成凝胶。这与 Voronkov[17]提出的环化反应是缩聚产物衍化成为可溶性多面体晶型低聚物、无定型低聚物或高聚物等不同物理存在状态的重要因素相符合。
基于有机取代基对水解-缩聚反应和产物结构衍化的影响,不同有机修饰硅氧烷其溶胶-凝胶产物可能具有不同的物理存在状态。Loy 等[18]在系统研究有机取代基对体系凝胶行为的影响时发现,不同取代基的硅氧烷水解有形成凝胶或者分相形成沉淀两种趋势,这与取代基的链长、空间位阻有关:含有链长较短及位阻较小以基团如甲基、乙烯基、氯甲基的前驱体水解-缩聚反应较快,在产物析出之前已经充分交联,易于形成凝胶。有趣的是含有如十八烷基、十二烷基长链基团的前驱体也容易形成凝胶,他们认为这是长链烷基之间的自组织行为使然。而介于两者之间的取代基则容易导致产物以油或者脂的形式存在。
值得注意的是,疏水性有机修饰的前驱体因为有机基团的存在造成水解产物产生明显的双亲性,这一特性会导致产物自组织成有序结构。这种作用在含有较长疏水有机取代基修饰的前驱体表现得更为突出。Matĕjka 等[8]在研究中发现,倍半硅氧烷无机骨架和侧链有机链的不相容性导致微区相分离发生,有机侧链在紧密的网络微区中自发组织产生胶束结构的有序排布。有机链越长,有序性越高,最多的组织排布发生在聚氧化乙烯(PEO)取代的聚硅氧烷中。最近,Hu等[19]借助荧光探针技术在线跟踪了有机修饰硅氧烷甲基丙烯酰氧丙基三甲氧基硅烷(MAPTMS)在本体相中的水解- 缩聚过程。依据检测过程中探针荧光发射光谱具有振荡行为提出,这一前驱体的水解产物具有两亲性质,在本体相中可以依据水量多少而形成油包水型或水包油型胶束结构的簇集体。这说明有机修饰前驱体的溶胶-凝胶过程中有机基团之间存在超分子相互作用是影响产物形态分布的不可忽视的重要原因。
有机修饰对水解-缩聚反应速度和结构衍化的影响必然导致最终产物的性质差异。除了宏观的表征手段之外,光物理技术是在分子水平上研究材料性质和结构的有力手段[12]。Baker[20]利用芘荧光分子研究了取代基分别为甲基、二甲基、乙基、二乙基、辛基和烯丙基有机修饰硅酸的极性。荧光光谱得到的信息证明了随着有机基团的链长增加,得到的材料的非极性越强。Qian[21]利用溶胶-凝胶过程将荧光物质香豆素440(7- 氨基-4-甲基-1-苯并吡喃-2-酮)包埋于有机修饰硅酸中,研究了引起光谱位移的基质效应和机理。荧光光谱信息表明香豆素440能够感受到较强的极性,说明体系中含有剩余的硅羟基和水,而在 MTES 和VTES 制备的材料中荧光最大发射波长位移微小,说明材料致密,只含有Si-O-Si网络结构和甲基或乙基侧基。
以上研究都表明有机取代基对前驱体的溶胶-凝胶过程具有较强的影响。目前,设计合成的有机修饰硅氧烷种类很多,对溶胶-凝胶过程的修饰行为各不相同。以下简单介绍几类具有典型结构特点的有机取代前驱体对溶胶-凝胶过程修饰行为。
1.1 桥联型前驱体
桥联型硅氧烷作为前驱体经由溶胶-凝胶过程制备的桥联聚倍半硅氧烷是有机-无机杂料中具有特殊性能的一类新型杂化材料[22]。桥联硅氧烷单体有机连接臂可以是柔性烷基链,也可以是不饱和结构的刚性链。桥联硅氧烷在较低浓度水解时即可导致枝化的网络快速生长,形成凝胶。有机连接臂的长度、刚性、取代基位置等因素可从分子水平上影响材料的结构[23]。连接臂是较短的烷基链,得到的聚合物具有多孔结构;连接臂较长,则柔性增加,孔道结构容易塌陷;刚性的共轭结构则会导致材料多孔性。
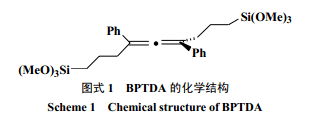
桥联型硅氧烷前驱体中含有特殊的有机连接臂会为水解缩聚反应过程分子自组装行为提供可能。Muramatsu[24]利用含有联苯基连接臂的桥型前驱体[1,4-二(5-三甲氧基硅烷基-1-戊氧基)二苯],经由固态反应制得了具有高度有序性材料。固体反应避免了溶胶的形成,从而削弱了溶剂对材料无规则形成的驱动作用。前驱体本身的晶型结构成为自组织行为的重要因素。固体中分子的结晶形态表面上不能在反应过程中保存,但是可以作为模板或者支架使聚合产物定向生长。取代基为联苯基、炔基苯基等刚性连接臂都具有上述类似自组织的特性。Cerveau[25]等发现一种具有旋拧线性结构硅氧烷桥联前驱体[1,3-二(苯基三甲氧基硅烷基)-1,3-二苯丙二烯,BPTDA,图式[1],其溶胶-凝胶过程具有纳米尺度上的自组织行为。前驱体含有扭曲的核心和柔性的侧链扩展了使体系能够有序排布的刚性连接臂范畴。有机连接臂之间通过范德华力、π-π堆积等弱的相互作用力诱导了材料结构的有序性使产物具有各向异性。
1.2 含疏水长链的前驱体
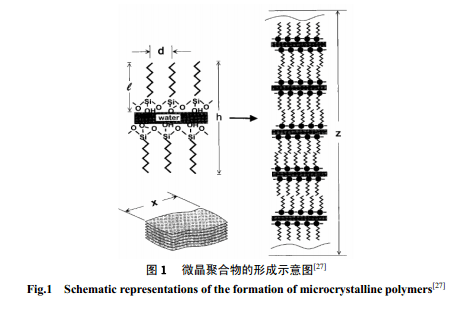
长链烷基取代硅氧烷类似表面活性剂的两亲性质和界面行为引起了人们极大的兴趣[26]。Parikh等[27]提出在膜-固体基质界面间有超薄的物理吸附水存在时形成的膜有序度最高。形成机制涉及到长链硅氧烷水解产物即[n-CnH2n+2-Si(OH)3]在准二维方向上进行的自组装。水解初级阶段较低的水解产物分子在膜-水界面进行类似表面活性剂形成 Langmiur 膜一样地侧向移动而相互靠近,形成覆盖面较小的膜层。随水解产物增加而形成高覆盖面积的单分子膜时,膜层中的水解产物经过硅羟基之间交联形成 Si-O-Si 键。除了在界面上形成 Langmiur 膜的二维结构,基于二维单分子自组装体的周期性模拟,可以设想 n-CnH2n+2-Si(OH)3 在本体相中能够形成高度有序的三维相结构。Parikh 等[27]通过研究[C18-Si(OH)
3]的有序排列组装形成层状微晶高聚物(图1) 证实了以上的预想。两亲的硅氧烷分子在气液界面上进行水解、缩聚,因气液界面间的惰性气体分子对两亲的硅氧烷分子的作用较弱,在此条件下两亲硅氧烷分子在气液界面的有序排列是靠分子自然排布而成的,避免了液固界面上的表面基团反应或基质的引入导致溶液中分子的重排。研究这类硅氧烷在气液界面上的二维溶胶-凝胶过程有助于深入理解前驱体的水解-缩聚反应机制及反应条件的作用原理。Vidon[28]利用π-Α曲线结合Brewster 角显微镜研究了十八烷基硅氧烷(C18TMS)在pH为1.4~12.8 范围内在水/气界面上形成单分子膜的过程,结果表明,前驱体无论是在酸性底相还是碱性底相中水解缩合形成的高聚物都为线性结构,归因于十八烷基相互间的位阻作用使缩聚产物定向生成,疏水基团有序分布在缩聚链的两侧。Britt 等[29]研究了十八烷基三甲氧基硅烷(OTMS)和十八烷基二甲基甲氧基硅烷(OMMS)在气液界面上的质子化、水解和缩合反应的动力学过程。表面压变化为常数说明硅氧烷中的三个甲氧基是分步进行质子化和水解的。膜层的延展时间、硅烷的堆积密度、底相pH都会影响到二维的水解缩聚过程。
1.3 含有活性基团的前驱体
有机修饰前驱体中含有功能化基团,可以赋予溶胶-凝胶材料独特的性能。例如,前驱体中含有环氧基可以开环聚合,具有质子传递行为[30];氨基的存在为溶胶-凝胶材料键合生物分子提供了方便[31];巯基的存在赋予材料具有和重金属的特异性结合功能[32];含有丙烯基或氨基等活性基团的前驱体可作为偶联剂增强有机相和无机相之间的复合程度[33],Schmidt 等[34]认为在无机网络中引入的有机组分主要起到两个方面的作用:网络性质修饰或作为网络结构的一部分。如果有机基团在整个溶胶-凝胶过程中没有发生化学反应,即“死基”,只是部分阻断了Si-O-Si 三维网络结构的形成,可以认为起到了网络修饰的作用。如果是活性基团则可以参与网络结构的形成。Mori 等[35]合成了含有多羟基的硅氧烷 N,N-二(2,3-二羟丙基)氨基丙基三乙氧基硅烷,此前驱体在酸性条件下经过水解-缩聚过程形成了平均粒径为 2.7nm 的颗粒。体系中没有形成大的团簇是因为烷基链功能化基团和硅原子之间通过共价键结合,所以颗粒的周围是高密度羟基功能化烷基链,阻碍了粒子之间的相互接触。同时,Mori 等发现连接在烷基链上的羟基可以化学键合到水解产物的硅羟基上,驱动了内部环化的产生,得到颗粒内部结构为Si-O-Si 和Si-O-C 组成的含有12~18 个原子的环状结构。对于含有可聚合功能基的前驱体而言,在溶胶-凝胶过程中通过光、热、或引发剂引发有机端聚合,因此导致有机网络和无机网络同步形成,而且存在着相互竞争[36]。Davis[37]以GLYMO 为前驱体制备有机-无机杂化材料,溶胶-凝胶过程以二乙三胺为催化剂催化环氧基开环聚合。结果证明,无机缩合和有机聚合反应同步进行,在二乙三胺浓度很低的情况下,有机聚合度较低,无机网络很快形成;高浓度的二乙三胺导致有机网络的快速形成,但是发现无机网络的发展缓慢。说明有机网络和无机网络的形成存在着相互作用。
1.4 键合高聚物的前驱体
高聚物特殊结构特点导致键合高聚物的前驱体水解-缩合过程的特殊性。Matĕjka 等[8]通过控制实验条件,使PEO 链节形成晶体,导致水解缩合产物在溶胶-凝胶过程中可能发生自组织行为。他们认为 PEO 键合前驱体溶胶-凝胶中间体在熔融态时可以形成球形胶束结构。温度降低时,分子量为2000 的PEO 链段可以完全舒展。而分子量为 5000 的PEO 链段可以折叠,形成的聚硅倍半氧烷微区晶体可能是部分互穿平行的结构,或者扭曲的双层结构。PEO 结晶行为驱动了聚硅倍半氧烷笼型结构排布成为层状结构。
将嵌段共聚物键合到硅氧烷前驱体上,可得到具有新结构特征的杂化材料。Mellon 等[38]通过可逆加成- 裂解链转移(RAFT)方法控制合成了聚甲基丙烯酸甲酯/ 聚甲基丙烯酰氧丙基三甲氧基硅烷[P(MMA-b-MPS)] 嵌段共聚物。P(MMA-b-MPS) 作为溶胶-凝胶过程的前驱体,他们期望通过改变反应条件得到具有可控性构造的嵌段共聚物,使其在溶胶-凝胶材料形成的过程中能够充分发挥其自组装的特性,这一研究思想将为结构型纳米材料的制备提供一个新途径。
1.5 其它含有特殊构型的前驱体
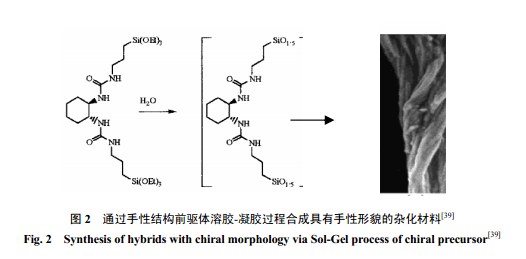
随着有机-无机杂化材料的广泛应用,人们别具匠心地设计了很多结构新颖的前驱体。Moreau等[39]设计合成了一种含有手性结构酰胺功能基团的桥型前驱体。他们利用这种前驱体经过溶胶-凝胶过程首次制备了具有手性形貌的杂化材料(图2) 。就像构建 DNA的双螺旋结构一样,氢键作用是材料自组织行为的驱动力,在反应过程中手性单体或缩合的低聚物之间的氢键弱相互作用可以导向Si-O-Si 键的定向生成。这类具有手性形貌和性质的杂化材料在非均相手性催化领域可能有重要的应用价值。Barboiu等[40]利用含有冠醚大环受体的前驱体设计合成了具有超分子结构的有机-无机杂化材料,有机基团之间的氢键作用在溶胶-凝胶过程中导致有机凝胶的定向生成。形成的有序性固体膜中的冠醚环对 Na+具有识别作用,基于这种离子识别和传输作用可作为三磷酸腺苷的离子驱动泵。这种特殊结构前驱体的研究结果展示了一个功能化有机硅氧烷通过自组装构建有序结构器件的范例。
2 外源性有机物对溶胶-凝胶过程的影响
除了对前驱体进行内源性修饰之外,外源性有机化合物引入到溶胶-凝胶体系也可以影响溶胶-凝胶过程。外源性有机物引入方式相对于内源性有机基团键合到前驱体更为简单灵活,使外源性有机物对溶胶-凝胶过程的修饰行为也变得异常复杂。下面根据外源性有机物在材料形成过程所起的作用简要介绍。
2.1 干燥控制添加剂
在溶胶- 凝胶的干燥过程中孔径变化产生的压力或溶剂挥发产生的毛细作用使凝胶块体产生裂纹或碎片,解决这一难题的有效方法之一是在材料的溶胶阶段加入干燥控制化学添加剂(DCCA)[41]。如甲酰胺、N,N-二甲基甲酰胺、乙腈、草酸等。DCCA 的作用机理较为复杂,一种看法是,DCCA的加入使材料具有孔径分布较窄的大孔结构,从而减弱了毛细作用,另一种看法是,DCCA 和硅酸表面上的羟基形成氢键可以阻止水分子和 Si-OH作用,且空隙中充满 DCCA 比充满水的表面张力小,从而使水分子容易挥发[42]。Viart 等[43]利用测试 pH和FT-IR 研究甲酰胺对 TEOS 溶胶-凝胶过程的作用机制时发现,没有甲酰胺存在时,前驱体缩合易生成线性产物,交联度较低;甲酰胺的存在使前驱体易生成小于6个硅原子的低聚物,在这些小的低聚物基础上进行再缩合增大了凝胶网络的交联度,使SiO2 网络结构趋向玻璃化。Lenza 等[44]在研究某些 DCCA 对溶胶-凝胶膜材料结构的影响时发现,DCCA 的存在下,较高温度制备的材料表面无裂纹的块体结构和介观尺度的大孔隙结构材料,且材料存在大量的自由硅羟基。
2.2 水解-缩聚反应调节剂
某些有机添加物可以对硅氧烷的溶胶- 凝胶速度施加影响。如对含有可聚合的环氧基前驱体(GLYMO)而言,1-甲基咪唑(MI)、3-氨丙基三乙氧基硅烷(APTES)和三氟化硼乙醚络合物[BF3(OEt)2]对其水解-缩合反应都会产生影响[37]。MI能够有效催化环氧基开环,但不直接影响网络结构的形成;APTES中的氨基不仅具有催化溶胶-凝胶过程作用,而且因分子中烷氧基存在,可参与凝胶网络的形成。BF3(OEt)2 具有有效提高无机端的交联度和聚氧化乙撑衍生物形成的双重功能。
值得关注的是,基于和前驱体分子中有机基团之间的相互作用,某些有机物可以对含有较长疏水链修饰的前驱体在气/液界面上的水解-缩聚反应产生影响。Park 等[45]研究了酸性表面活性剂全氟代十四烷酸(PFTDA) 对气/ 液界面上形成的十八烷基三甲氧基硅烷(ODTMS)单分子膜胶凝行为的影响,结果表明加入2(mol)% PFTDA 即可加速 ODTMS 凝胶形成,他们认为 PFTDA在单分子层中快速扩散,质子很快传播到反应活性位点甲氧基附近,导致水解反应部位酸度很高,从而催化了ODTMS 的水解。通过Cd2+与PFTDA形成络合物从而减弱了 PFTDA催化活性的实验结果证实上述推测。Wang等[46]利用10,12-二炔基-二十五烷酸(PDA)和它的衍生物N-(11-O-α-D-吡喃葡糖-3,6,9-三氧杂)十一烷基-10,12-二炔基-二十五烷酸(PDTMC)以及十八烷基三甲氧基硅烷(ODTMS)构成Langmuir 单分子膜(图3) ,混合物之间存在两种作用力:一方面较长碳链之间的疏水相互作用;另外ODTMS 单体水解产物硅羟基和 PDA 分子中羟基以及 PDTM 分子中酰氨基之间存在氢键作用。因此适当的 pH 条件下,混合物互溶没有相分离发生时,彼此之间产生了立体位阻效应,即“空间隔离效应”,导致 PDA/PDTM的光聚合反应和 ODTMS 的缩和反应能够相互影响。利用这一效应来调节硅氧烷溶胶-凝胶反应的有机物还有硬脂酸甲酯[47]等一些惰性有机物。

2.3 模板及结构导向剂
有机物对溶胶-凝胶过程中的水解-缩合产物往往表现出模板或结构导向作用。利用有机物作为模板剂合成各种孔道结构的溶胶-凝胶材料的相关工作已经有大量的文献报道[48-50]。模板可以是以共价键维系特异形貌的具有不同空间结构的硬模板,如高分子聚合物[51]、水凝胶[52]或有机凝胶[53];也可以是通过分子间或分子内的弱的相互作用力而形成的分子自组装体或超分子模板[54-55]。这里,笔者着重介绍有关在溶胶-凝胶过程中有机物发挥模板作用或结构导向剂作用的机理研究。Beck等[50]以表面活性剂形成的液晶为模板合成了一系列 MCM-41 型介孔分子筛,并且提出了表面活性剂簇集形成六角形的棒状胶束导致材料特殊形貌形成的机理。实验证明,材料形成的结构和孔径与表面活性剂的链长及溶解性密切相关。Sahu 等[56]利用香豆素 480 跟踪了表面活性剂CTAB 存在下,溶胶-凝胶过程中溶剂化动力学过程时发现,CTAB 的极性头基和硅羟基之间的相互作用使包埋在其中的水运动受限,探针分子的各向异性值充分证明了疏水核心的存在。最近,Zhang 等[57]以一种类似“蜥蜴”型的分子作为模板设计合成了功能化的介孔二氧化硅材料。模板分子为两亲物,阳离子头基共价结合于可缩合的硅氧烷基团;中间为欲赋予材料的功能基团;尾部为疏水性的烷基链,且在分子固定后可在酸性条件下水解实现“剪切”。在“剪切”前的疏水尾可以保证前驱体具有自组装性质,在固定后剪切使功能基释放裸露于材料孔道中。该方法为具有特殊分子结构新型模板的设计开辟了一条新途径。Chen等[58]以非离子三元嵌断共聚物聚氧化乙烯-聚氧化丙稀-聚氧化乙烯(EO20 PO70 EO20)和阴离子表面活性剂为结构导向剂经由 TEOS 溶胶-凝胶过程制备具有双连续立方结构的介孔二氧化硅材料,结果发现,调节共聚物和表面活性剂的含量可以提高溶胶-凝胶材料的有序性,说明是二者共同组织的胶束簇集体发挥了模板的作用。Shinkai等[59]]首次利用不含氨基和静电荷的有机分子和苯胺类有机溶剂形成的有机凝胶为结构导向剂经由 TEOS 溶胶-凝胶过程合成了纤维状二氧化硅材料。不同于以往的氢键作用,苯胺分子和胶凝剂分子之间的疏水相互作用以及 π-π 弱相互作用是模板结构转录到二氧化硅材料中的驱动力。
类似于生物的矿化作用,在溶胶-凝胶过程中有机物和无机物能够实现在分子水平上的穿插复合本身就意味着相互间的导向作用。孙继红等[60]研究了聚氧乙烯(PEO)-聚氧丙醚(PPO)-聚氧乙烯醚(PEO)(简称 EPE)三元嵌段共聚物作为改性剂对溶胶-凝胶过程的修饰行为。EPE 首先与体系中微量水相互作用,通过影响 TEOS 的水解缩聚反应,改变了Si 的配位环境,EPE 疏水基和亲水基的存在使得基本结构单元形成弯曲度较大的链状结构,从而导致SiO2 溶胶的网络结构。Shchipunov 等[61]利用各种天然多糖大分子和可溶性前驱体(TEOS 的低聚物)合成了新一类块体硅酸生物材料,认为大分子结构中的羟基和硅醇之间的氢键作用或形成共价键为二氧化硅颗粒的生长提供了“晶核”,而且在整个溶胶-凝胶过程中控制颗粒的生成位点。Tamaki 等[62]利用聚苯乙烯和苯基取代硅氧烷合成了均匀的凝胶高分子材料,认为高聚物和苯基之间的 π-π 弱相互作用使得聚苯乙烯和硅酸凝胶均匀分散,而聚苯乙烯和其它不含苯基前驱体在同样条件下制备的杂化材料表现出明显的不均一性。这一结果说明利用芳香类高聚物结合硅氧烷的溶胶-凝胶过程制备材料时,要充分考虑芳环之间的π-π 弱相互作用。
3 展望
到目前为止,对于有机组分在分子水平上对硅氧烷水解-缩合反应过程的机理研究不是很多,因此有关该领域存在较大的研究空间。首先,结合当今各种先进的实验分析手段和检测技术,如液相/固相29Si NMR、SEM和TEM、SAXRD、AFM、凝胶色谱,特别是采用多种原位技术跟踪溶胶-凝胶过程十分必要。通过29Si NMR 在线检测可以获得水解-缩聚反应的动力学过程;通过Raman 、IR光谱在线检测也可以获得水解-缩聚反应过程中物种变化的信息。另外,由于此研究体系具有不均性特点,因此,有关胶体与界面研究的方法如激光光散射、AFM、BET 、表面张力测定、光物理技术等是研究溶胶-凝胶过程的强有力手段。例如利用荧光探针技术进行在线跟踪溶胶-凝胶过程能够提供由于物种分布变化所引起微环境变化的重要信息。除上述有关实验手段外,作为实验的有力补充和证明手段,利用分子模拟方法也有助于人们从理论上认识溶胶-凝胶动力学过程[63-64]。再有,以特殊结构化合物作为“模型” 研究对象有助于对复杂体系机理的理解。通过分子设计合成具有“预想” 结构的目标产物,可以推测和验证无机组分与有机组分相互作用的本质。我们相信,研究有机修饰对硅氧烷溶胶-凝胶过程的影响无论对于理论研究和实际应用均具有重要的意义。
参考文献
[1] L L Hench, J K West. Chem. Rev., 1990, 90: 33~72.
[2] G Schottner. Chem. Mater., 2001, 13: 3422~3435.
[3] A V Cunliffe, S Evans, D A Tod et al. Int. J. Adhes. Adhes., 2001, 21(4): 287~296.
[4] B Alonso, D Massiot, F Babonneau et al. Chem. Mater., 2005, 17: 3172~3180.
[5] R J Hook. J. Non-Cryst. Solids, 1996, 195: 1~15.
[6] P Innocenzi , G Brusatin. Chem. Mater., 2000, 12: 3726~3732.
[7] M Gnyba, M Keränen, M Kozanecki et al. Opto-Electro. Rev., 2002,10(2): 137~143.
[8] L Matĕjka, O Dukh, Drahomíra et al. Macromolecules, 2001, 34: 6904~6914.
[9] Y Wei, D Jin, D J Brennan et al. Chem. Mater., 1998, 10: 769~772.
[10] K Piana, U Schubert. Chem. Mater., 1994, 6: 1504~1508.
[11] Y Kaneko, N Iyi, K Kurashima et al. Chem. Mater., 2004, 16: 3417~3423.
[12] T K Tucker, J D Brennan. Chem. Mater., 2001, 13: 3331~3350.
[13] S Yano, K Iwata, K Kurita. Mater. Sci. <st1:country-region w:st="on">Eng. C, 1998, 6: 75~90.
[14] R Winter, D W Hua, X Song et al. J. Phys. Chem., 1990, 94: 2706~2113.
[15] C A Fyfe, P P Areca. J. Phys. Chem. B, 1997, 101: 9504~9509.
[16] Y Sugahara, T Inoue, K Kuroda. J. Mater. Chem., 1997, 7(1): 53~59.
[17] M G Voronkov, V I Lavrent’yev. Top. Curr. Chem., 1982, 102: 199~236.
[18] D A Loy, B M Baugher, C R Baugher et al. Chem. Mater., 2000, 12: 3624~3632.
[19] D D Hu, C C Barghorn, M Feuillade et al. J. Phys. Chem. B, 2005, 109: 15214~15220.
[20] G A Baker, S Pandey, E P Maziarz et al. J. Sol-Gel Sci. and Technol., 1999, 15: 37~48.
[21] G Qian, Z Yang, C L Yang, J. Appl. Phys., 2000, 88(5): 2503~2508.
[22] D A Loy, K J Shea, Chem. Rev., 1995, 95: 1431~1442.
[23] 刘丰袆, 符连社, 王俊 等. 化学通报, 2003, (6): 382~387.
[24] H Muramatsu, R Corriu, B Boury. J. Am. Chem. Soc., 2003, 125: 854~855.
[25] G Cerveau, R J P Corriu, E Framery et al. Chem. Mater., 2004, 16: 3794~3799.
[26] T Koga, M Morita, H Ishida et al. Langmuir, 2005, 21: 905~910.
[27] A N Parikh, M A Schivley, E Koo et al. J. Am. Chem. Soc., 1997, 119: 3135~3143.
[28] S Vidon, R M Leblanc. J. Phys. Chem. B, 1998, 102: 1279~1286.
[29] D W Britt, V Hlady. Langmuir, 1999, 15: 1770~1776.
[30] M Popall, H Durand. Electrochimica Acta, 1992, 37: 1593~1597.
[31] F Zhang, M P Srinivasan. Langmuir, 2004, 20: 2309~2314
[32] J B Jia, B Q Wang, A Q Wu et al. Anal. Chem., 2002, 74: 2217~2223.
[33] M Abdelmouleh, S Boufi, A B Salah. Langmuir, 2002, 18: 3203~3208.
[34] X C Li, T A Ling. J. Non-Cryst. Solids, 1996, 204: 235~242.
[35] H Mori, M G Lanzendo1örfer, A H E Mu1Üller. Macromolecules, 2004, 37: 5228~5238.
[36] P Innocenzi, G Brusatin. Chem. Mater., 2000, 12: 3726~3732.
[37] S R Davis, A R Brough, A Atkinson. J. Non-Cryst. Solids, 2003, 315: 197~205.
[38] V Mellon, D Rinaldi, E B Lami et al. Macromolecules, 2005, 38: 1591~1598.
[39] J J E Moreau, L Vellutini, M Wong Chi Man et al. J. Am. Chem. Soc., 2001, 123: 1509~1510.
[40] M Barboiu, S Cerneaux, A Lee et al. J. Am. Chem. Soc., 2004, 126: 3545~3550.
[41] C J Brinker, G W Scherer. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing. CA: Academic Press, 1990.
[42] D Phillips, P Brien, S Roberts. Sol-Gel Materials: Chemistry and Applications. <st1:country-region w:st="on">UK: Taylor & Francis Books Ltd, 2003.
[43] N Viart, J L Rehspringer. J. Non-Cryst. Solids, 1996, 195: 223~231.
[44] R F S Lenza, W L Vasconcelos. J. Non-Cryst. Solids, 2003, 330: 216~225.
[45] H K Park, T H Ha, K Kim. Langmuir, 2004, 20: 4851~4858.
[46] S P Wang, S Vidon, R M Leblanc. J. Colloid Interface Sci., 1998, 207: 303~308.
[47] D W Britt, V Hlady. J. Phys. Chem. B, 1999, 103: 2749~2754.
[48] C T Kresge, M E Leonowicz, W J Roth et al. Nature, 1992, 359: 710~712.
[49] B Hatton, K Landskron, W Whitnall et al. Acc. Chem. Res., 2005, 38: 305~312.
[50] J S Beck, J C Vartuli, W J Roth et al. J. Am. Chem. Soc., 1992, 114:10834~10843.
[51] T Amatani, K Nakanishi, K Hirao et al. Chem. Mater., 2005, 17: 2114~2119.
[52] H P Hentze, E Krämer, B Berton et al. Macromolecules, 1999, 32: 5803~5809.
[53] J H Jung, Y Ono, K Sakurai et al. J. Am. Chem. Soc., 2000, 122: 8648~8653.
[54] P T Tanev, Y Liang, T J Pinnavaria. J. Am. Chem. Soc., 1997, 119: 8616~8624.
[55] M Mureseanu, A Galarneau, G Renard et al. Langmuir, 2005, 21: 4648~4655.
[56] K Sahu, D Roy, S K Mondal et al. J. Phys. Chem. B, 2004, 108: 11971~11975.
[57] Q M Zhang, K Ariga, A Okabe et al. J. Am. Chem. Soc., 2004, 126: 988~989.
[58] D H Chen, Z Li, C Z Yu et al. Chem. Mater., 2005, 17: 3228~3234.
[59] A Friggeri, O Gronwald, K J C van Bommel et al. Chem. Commun., 2001, 2434~2435.
[60] 孙继红, 巩雁军, 王树国 等. 无机化学学报, 2000, 16(1): 131~135.
[61] Y A Shchipunov, T Yu Karpenko. Langmuir, 2004, 20: 3882~3887.
[62] R Tamaki, K Samura, Y Chujo et al. Chem. Commun., 1998, 1131~1132.
[63] J Sÿef čík, S E Rankin. J. Phys. Chem. B, 2003, 107: 52~60.
[64] H C Lay, M J S Spencer, E J. Evans et al. J. Phys. Chem. B , 2003, 107: 9681~9691.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号