有机合成中的复分解方法的发展
- 2012-06-26
- 专题
1 引言
复分解(Metathesis)这个词来源于希腊的meta(变化)和thesis(位置) ,是指两种物质的部分交换。在反应 AB+CD→AC+BD 中,B 已经与C 交换了位置。式1 示出了烯烃交换反应,通过两个原料烯烃之间的碳烯(亚烷基)交换,形成了两个新的烯烃。

复分解催化剂已经发展成为有机合成中的极其强有力和多才多艺的工具,它所导致的合成变换令人叫绝。
下面来叙述由肖夫所揭示的复分解机理和随后由施罗克与格拉布发现的复分解催化剂及其应用。
2 合成方法论的诺贝尔奖
尽管迄今为止,在惊人数量的有机分子中,还只有很少的一部分为合成化学家们所探索,然而,就是这些已经给我们人类提供了许多新的药物、新的农业化学品和新材料等,如果没有这些,人类将无法生存。对此的进一步探索必将对人类带来更巨大的利益,潜力是惊人的。然而,这种进展要求必须要开发新的选择性合成方法!
自一个世纪前诺贝尔奖建立以来,有机化学的合成方法方面已经获得了5 次诺贝尔化学奖。所有这些都包括了碳-碳键的构建及其化学。对于碳-碳键及其在有机合成中的重要性是不言而喻的。1912 年的诺贝尔化学奖授予了 Grignard 和 Sabatier,以表彰他们在确认格氏试剂对于构建分子中碳-碳键的形成的重要性以及在不饱和化合物的催化氢化反应中使用金属所做出的巨大贡献。1950 年,诺贝尔化学奖授予了 Diels-Alder 反应的发明人,以表彰他们以最简单的方式重组碳-碳双键而形成新的碳-碳键(2个单键和一个双键一个)和一个六元环。1979 年,诺贝尔化学奖分别授予了 Brown 和 Wittig 以表彰他们在碳-碳键化学(不饱和碳-碳键的硼氢化反应及其化学)和其形成(Wittig 反应)方面的贡献。2001年,碳-碳键化学因 Knowles和野依良治(双键还原) 以及Sharpless(双键氧化)对不对称催化剂方面的工作而再次获奖。今年,复分解方法(催化打断碳-碳双键和形成)的发展再次获此殊荣。
3 烯烃复分解反应的发现
催化复分解反应是随上世纪 50 年代由齐格勒(Ziegler,1963 年诺贝尔化学奖)在乙烯聚合反应中的观察而在工业中发现的。尽管在一系列专利中报道了新的过程,但是对其机理并不了解。其中的一个专利是由杜邦公司的 Eleuterio 在 1957 年公布的有关不饱和聚合物的形成。此聚合物是由高张力的原料降冰片烯加到负载在与氢化锂铝结合的氧化铝的氧化钼上而获得的[1a]。同一年,申请的另一个有关烯烃歧化反应的专利,即用负载于氧化铝上的三异丁基铝和氧化钼处理使丙烯转换成乙烯和丁烯,揭示了另一个似乎是新的转换[1b,c]。
1966 年,那塔(Natta,1963 年诺贝尔化学奖)及其合作者指出,六氯化钨与三乙基铝或二乙 基氯化铝的结合使环庚烯、环辛烯和环癸烯聚合[2]。下一年,Calderon 及其合作者报道了通过采用六氯化钨和乙基氯化铝的混合物为引发剂把上述发现延伸到其他的环烯烃[3a,b]。Calderon 认为,环烯烃聚合成多烯单体和脂环烯的歧化是同样类型的反应,建议称其为“烯烃复分解反应”[3d]。他们的结果引起了其他科学家对把此反应引入有机和金属有机反应中的潜力的兴趣。然而,对于复分解的机理的理解仍然还是一个谜。
4 复分解是如何进行的-肖夫机理
早期有几种对烯烃复分解机理的解释。首先,对于烯烃复分解是否有烷基或亚烷基的交换是有疑问的。Calderon 和 Mol用同位素标记的烯烃的实验证实,烯烃复分解中相互交换的基团是亚烷基[3a∼c]。但是,对于机理中的相互交换以及金属物种所起的作用起码还是臆测的。在那时流行的有关机理的几种说法是用 Calderon 的金属-配位环丁烯模型[3d]来解释亚烷基交换的。后来,又提出了Grubbs 的金属脂环戊烷模型[8a]。这些模型最初往往是不同的作者用不同的方法来描述的(式 2) 。
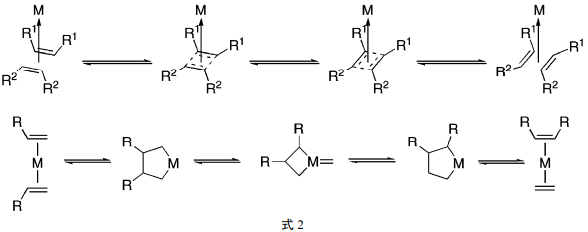
然而,直到法国石油研究所的肖夫对复分解机理的了解的努力以及 Fisher(1973 年诺贝尔化学奖)对于钨-碳烯复合物的合成、那塔关于通过由WCl6和AlEt3 的混合物催化的开环使环戊烯聚合、Banks 和Bailey 有关用负载在氧化铝上的W(CO)6催化使丙烯形成为乙烯和2-丁烯的报道之后,才提出了可行的机理。1971年,肖夫和他的学生Jean-Louis Herisson 发表了他们的复分解机理,式3 以修饰的形式示出了他们的机理。

式3 中,金属碳烯(亚甲基金属)作为催化剂,把两个端烯烃复分解为一个内烯烃(E和Z 异构体的混合物)和乙烯。复分解是可逆反应,但是,在这里,乙烯的移走使得反应完全了。
式3b 示出了肖夫催化循环[4] 。亚甲基金属(金属碳烯)与烯烃反应,形成了金属环丁烯中间体。然后,此中间体裂解产生了乙烯和新的金属碳烯。所形成的乙烯一个来自于催化剂的亚甲基和一个来自于原料烯烃。新的金属碳烯含有带有配体的金属(用[M]表示)和来自于底物烯烃的亚烷基。此亚烷基金属与一新的底物烯烃分子反应产生另一个金属环丁烯中间体。在正向反应的分解中,此中间体产生一个内烯烃产物和亚烷基金属。此亚烷基金属准备进入另一个催化循环。因此,催化循环中的每一步都包括了亚烷基交换-复分解。
肖夫与其合作者还提出了对此机理的实验证据,而这些证据不能用其它已经提出的机理来解释[4]。Grubbs、Katz、Schrock 和其他一些人的实验也支持了这个机理,因此,现在已经被普遍接受为复分解的机理。
肖夫的亚烷基金属引发复分解机理告诉我们,人们可以只需合成金属-亚烷基复合物并让它们作为催化剂来与烯烃进行复分解反应。[在那时已知的Fisher 型过渡金属碳烯复合物并不是复分解催化剂。
已经有许多科学家对催化剂的发展做出了贡献,其中Lappert的有关铑(I)复合物催化剂[5a]、Casey 的钨复合物[5b]的复分解和其他一些工作是重要的早期贡献。然而,这里的焦点在于由Schrock 和 Grubbs 在开发对于现代有机合成产生如此巨大影响的金属-亚烷基复合物方面的突破。
5 Schrock 对于第一个明确、有用的催化剂的创新
尽管许多科学家看好复分解的巨大合成潜力,但是要把它用到有机化学中却是很复杂的,因为传统的催化剂对于空气和湿气非常敏感而且会引起副反应以及其寿命相对较短。因此,要取得进展就要求要有可以鉴定的、相对稳定的长寿命催化剂,而且甚至其反应性要随所执行的任务“可调”。
杜邦公司的Schrock 在上世纪70 年代初,试着合成[Ta(CH2CMe3)5],并预期它将是一种稳定的化合物。他确实分离得到的了第一个稳定的金属-亚烷基复合物,[Ta(CH2CMe3)3(=CHCMe3)],它有高(V)的氧化态[6a]。
然后,Schrock 合成了包括第一个亚烷基复合物在内的其它的钽-亚烷基复合物,并用 X-线衍射和核磁共振进行了表征。他还发现,形成了金属环丁烯类。但是,在这些亚烷基复合物中没有一个可以催化烯烃的复分解[6b]。
1980年,Schrock 与其在麻省理工学院的研究小组报道了一种钽-亚烷基复合物[Ta(=CHC(CH3)3Cl(PMe3)(O-C(CH3)3)2]催化了顺-2-戊烯的复分解。此复合物能够起作用而其他的铌和钽的亚烷基复合物不起作用的原因在于,它存在有烷氧化物配体[6c,d]。
如上所述,在烯烃复分解中,钼和钨是活性最大的金属。Schrock 和他的研究小组加大了他们对寻找稳定分子亚烷基和这些金属亚烷基复合物的努力,最终得到了以通式[M(=CHMe2Ph)(=N-Ar)(OR)2](其中的R为大体积基团)表示的一系列钼和钨的亚烷基复合物。这些复合物是目前已经知道的活性最好的烯烃复分解催化剂(式4) [6e-h]。
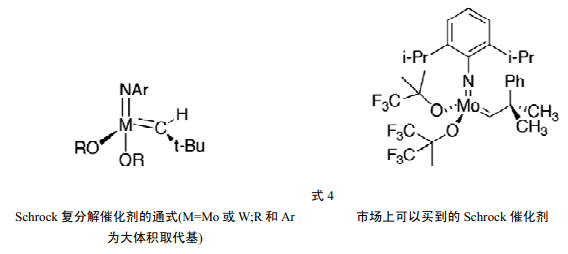
包括斯特拉斯堡的Osborn 和里昂的Basset 也以所报道的钨复合物对活性烯烃复分解催化剂做出了重要贡献。
Schrock 催化剂中最有效的是在1990 年报道的,这些催化剂的优点除了它们的活性之外,还在于它们是分子(没有添加剂)。Schrock 还与Hoveyda 一起开发了不对称催化复分解的手性催化剂。下面示出了一些应用Schrock催化剂的合成。
6 Grubbs 发现了实用的催化剂
Grubbs 早就对复分解反应感兴趣了,比如他就提出过金属环戊烯中间体的机理[8a]。自在上世纪 80 年代中期开始的对一些用后过渡金属盐制备的不太成功的催化剂的探索之后,Grubbs 及其合作者发现,三氯化钌甚至可以在水中使烯烃聚合[8b]。实际上,三氯化钌早已经被Natta用作环丁烯开环聚合的催化剂[7]。Grubbs假设此催化剂也是由金属碳烯机理来进行的。他们的结果引起了对于可在采用标准有机技术以及对宽范围官能团适用的催化剂的开发。
作为他们的开发工作的结果,Grubbs 和他的合作者在 1992 年报道了他们的第一个分子钌-碳烯复合物,它不仅对降冰片烯的聚合有活性,而且在存在质子溶剂时也是稳定的[8c]。复合物是亚乙烯基型的[RuCl2(PR3)(=CH-CH=CPh2)],其中的R=Ph(式5)。为了增加催化剂的反应活性,把苯基换成环己基(R=Cy)[8d,e],此变化产生了所期望的反应性、催化了无张力的烯烃聚合和诱导了与脂环烯烃的反应。它促进了许多与Schrock 钼基亚烷基复合物同样的反应,而且可以有更大的官能团和可以用标准的有机技术来处置[8f]。Noels 小组在1992 年也报道了环烯烃的Ru-催化开环复分解聚合(ROMP) 。
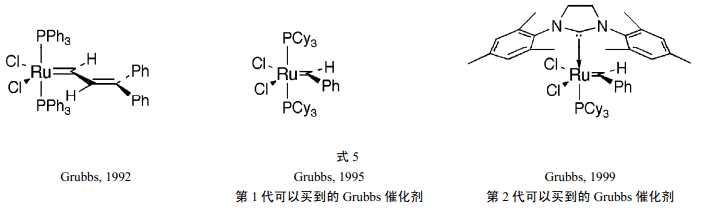
随着对需要的催化剂数量的增长,要求要有更有效的合成方法。为此,已经开发了一个亚苄基钌复合物的实用途径。1995 年,Grubbs 报道了新的分子催化剂[Ru(=CHPh)Cl2(PR3)2],R 为苯基或环己基(Cy)[8g,h]。它们的结构与亚烯基的很相近。R 为环己基的化合物已经作为第一代 Grubbs催化剂而商业化(式 5),由于其在空气中的稳定性和与大量基团的兼容性,此化合物仍是有机化学家们最广泛使用的复分解催化剂。
在大量的难以关环的反应中,对于在其合理负载量下以高产率得到产物而言,催化剂的寿命仍然是不够的,需要出现性能改进的催化剂。Grubbs领导的小组通过对机理的深入研究,得到了反应首先包括其一个膦的解离以产生反应性钌中间体的结论。为了加速解离,Grubbs把一个膦用环双氨基碳烯配体 A 来代替。Herrmann 早已经合成了这种有两个配体的钌复合物,但是它们的催化剂活性一般。Grubbs的催化剂仅含一个这种配体,加快了余下的一个膦的解离速度,从而加大了其复分解活性。1999 年 Nolan和 Fürstner与 Herrmann几乎同时发表了类似的结果。新的更高活性的催化剂被称为第二代Grubbs催化剂。
[RuCl2{C(N(mesityl)CH2)2}(Pcy3)(=CHPh)](式 5)是目前最广泛使用的交叉复分解反应的有效催化剂[8i] 。此新的钌催化剂具有更好的热稳定性,现在市场上也可以买到。
Grubbs 的成功也使得其他一些科学家如 Hoveyda、Hofmann、Grela和 Blechert等得到了改进钌基催化剂的灵感和目标。下面示出了一些采用 Grubbs催化剂的合成。
7 应用的多样性
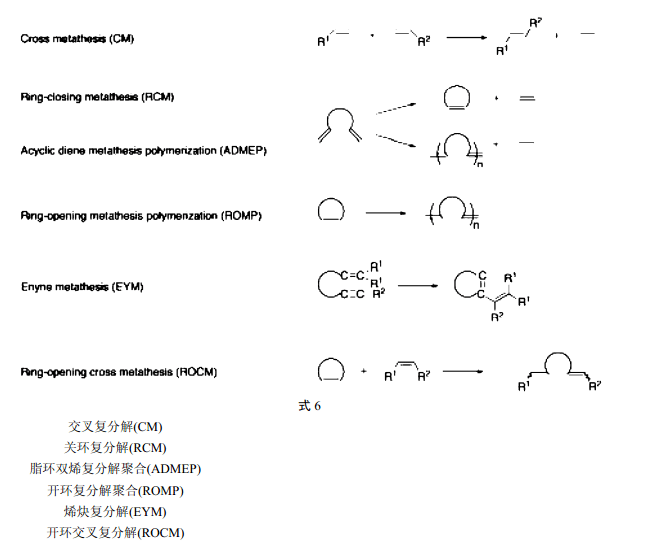
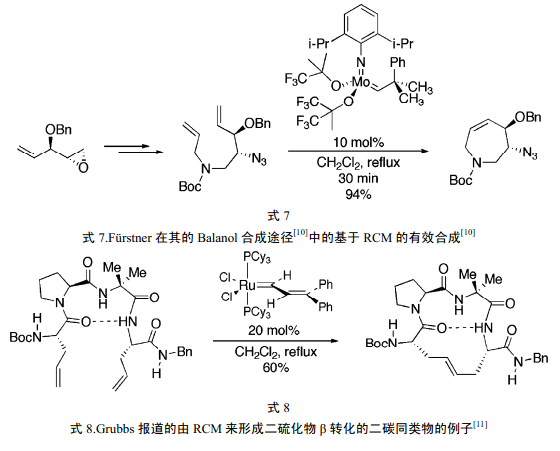
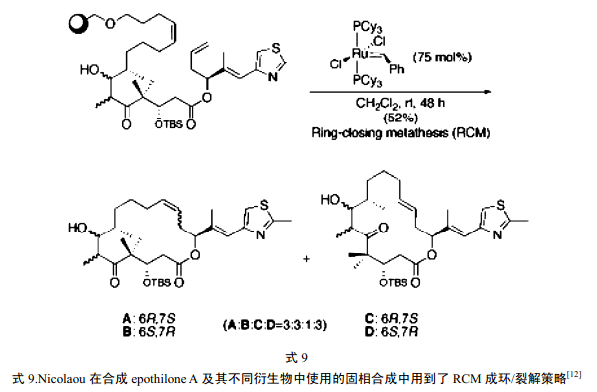
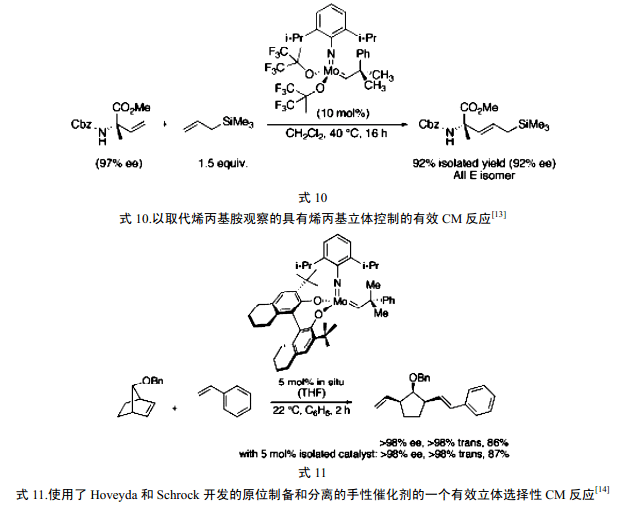
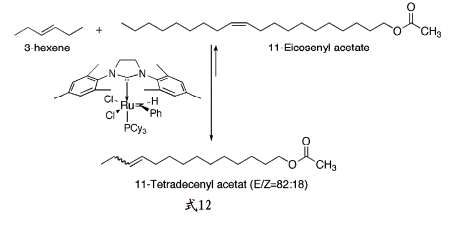
Grubbs 和 Schrock 催化剂给合成化学家提供了新的机会,它们之所以能在有机化学中得到广泛应用是得益于它们对大量官能团的包容性和效率的结合,而对 Grubbs 催化剂来说,还在于其容易在空气中处置[9]。式 6 示出了不同类型的复分解反应,随后的例子说明了新型催化剂的能力。
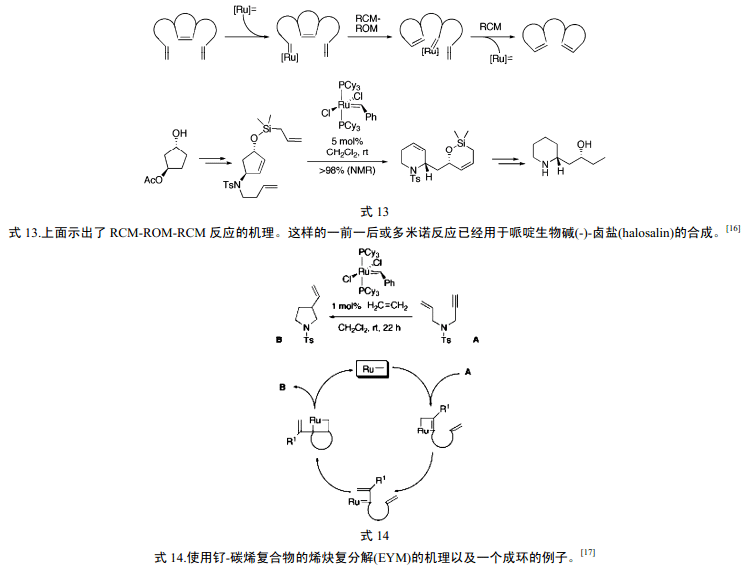
8 催化剂的进一步发展
催化剂的设计(或在某些情况下的再设计)留给人们巨大的余地,几乎没有一个月没有用于复 分解的新催化剂的揭示。许多这样的新体系促进和推动了构建高官能化复合分子的努力。复分解所显现的使转化成为有效的能力是其它方法所难以达到的,这种趋势必将持续下去。
9 结果与应用
强调诺贝尔奖得主的发现与改进对于科学研究和商品化合物的工业开发的巨大意义是十分重要的。考虑到 Grubbs 和 Schrock 催化剂的应用时间还很短,所取得的突破是非常惊人的。我们已经目睹了具有特殊性质的聚合物、聚合物添加剂、燃料和生物活性材料比如昆虫信息素、除草剂和药物的合成。
由于催化复分解缩短了合成路线,使得反应有更高的产率。这使得我们探索更多更新的有机分子成为可能,从而也可以对“绿色化学”做出贡献。
参考文献
[1] (a) Ger. Pat. 1 072 811 (1960) toH.S. Eleuterio, Chem. Abstr., 55 (1961) 16005; U.S. Pat. 3 074 918 (1063) to H.S. Eleuterio.
(b) H.S. Eleuterio, J. Mol. Cat., 65, 55 (1991) and references therein.
(c) R.L. Banks and G.C. Bailey, Ind. Eng. Chem., Prod. Res. Develop., 3, 170 (1964).
[2] G. Natta, G. Dall`Asta, I.W. Bassi, and G. Carella, Makrol. Chem. 91, 87 (1966).
[3] (a) N. Calderon, H.Y. Chen, and K.W. Scott, Tetrahedron Lett., 3327 (1967).
(b) N. Calderon, E.A. Ofstead, J.P. Ward, W.A. Judy , and K.W. Scott, J. Am. Chem. Soc. 90, 4133 (1968).
(c) J.C. Mol, J.A. Moulijn, and C. Boelhouwer, Chem. Commun., 633 (1968).
(d) N. Calderon, Acc. Chem. Res. 5, 127 (1972).
[4] J.-L. Hérisson and Y. Chauvin, Macromol. Chem., 141, 161 (1971). (b) J.-P. Soufflet, D. Commereuc and Y. Chauvin, 276, 169 (1973).
[5] (a) D.J. Cardin, M.J. Doyle and M.F. Lappert, J.Chem. Soc., 927 (1972).
(b) C.P. Casey and Burkhardt, J. Am. Chem. Soc., 96, 7808 (1974).
[6] (a) R.R. Schrock, J. Am. Chem. Soc., 96, 6796 (1974).
(b) C.D. Wood, S.J. McLain and R.R. Schrock, J. Am. Chem. Soc., 101, 3210 (1979).
(c) R.R. Schrock, S.M. Rocklage, J.H. Wengrovius, G. Rupprecht and J. Fellmann, J. Molec. Catal. 8, 73 (1980).
(d) S.M. Rocklage, J.D. Fellman, G.A. Rupprecht, L.W. Messerle and R.R. Schrock, J. Am. Chem. Soc., 103, 1440 (1981).
(e) J.S. Murdzek and R.R. Schrock., Organometallics, 6, 1373 (1987).
(f) R.R. Schrock, S.A. Krouse, K. Knoll, J. Feldman, J.S. Murdzek and D.C. Yang, J. Molec. Catal., 46, 243 (1988).
(g) R.R. Schrock, J.S. Murdzek, G.C. Barzan, J. Robbins, M. DiMare and M. O`Regan, J. Am. Chem. Soc., 112, 3875 (1990).
(h) C.G. Bazan, J.H. Oskam, H.-N. Cho, L.Y. Park and R. R. Schrock, J. Am. Chem.Soc. 113, 6899 (1991).
[7] G. Natta, G. Dall`Asta and L. Porri, Makromol. Chem., 81, 253 (1965).
[8] (a) R.H. Grubbs and T.K Brunck, J. Am. Chem. Soc., 94, 2538 (1972).
(b) B.M. Novak and R.H. Grubbs, J. Am. Chem. Soc., 110, 960 (1988).
(c) S.T. Nguyen, L.K. Johnsson, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc., 114, 3974 (1992).
(d) Z. Wu, S.T. Nguyen, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc., 117, 5503 (1995).
(e) S.T. Nguyen, R.H. Grubbs,J.W. Ziller, J. Am. Chem., 115, 9858 (1993).
(f) G.C. Fu, S.T. Nguyen, R.H. Grubbs, J. Am . Chem. Soc., 115, 9856 (1993).
(g) P. Schwab, M.B. France and J.W. Ziller, Angew. Chem.. Int. Ed. Engl., 34, 2039 (1995), Angew. Chem. 107, 2179 (1995).
(h) P. Schwab, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc. 118, 100 (1996).
(i) M. Scholl, T.M. Trnka, J.P. Morgan and R.H. Grubbs, Tetrahedron Lett.40, 2247 (1999).
[9] Handbook of Metathesis; R.H. Grubbs, Ed.,Wiley-VCH: New York, (2003).
[10] A. Fürstner and O.R. Thiel, J. Org. Chem. 65, 1738 (2000).
[11] S.J. Miller, H.E. Blackwell and R.H. Grubbs, 118, 9606 (1996).
[12] K.C. Nicolaou, N. Winssinger, J. Pastor, S. Ninkovic, F. Sarabia, Y. He, D. Vourloumis, Z. Yang, T. Li, P. Giannakakou and E. Hamel, Nature 387, 268 (1997).
[13] O. Brümmer, A. Rückert and S. Blechert, Chem. Eur. J., 3, 441 (1997).
[14] X. Teng, D. Cefalo, R.R. Schrock and A.H. Hoveyda., J. Am. Chem. Soc., 124, 10079 (2002).
[15] R.L. Pederson, I.M. Fellows, T.A. Ung, H. Ishihara and S.P. Hajela, Advanced Synthesis & Catalysis, 344, 728 (2002).
[16] R. Stragies and S. Blechert, Tetrahedron 55, 8179 (1999).
[17] M. Mori, N. Sakakibara and A.Kinoshita, J.Org.Chem., 63, 6082 (1998).Further reading The Age of the Molecule, N.Hall, Royal Society of Chemistry, London, 1999. Olefin metathesis: Big-Deal Reaction, C&EN, December 23, 2002, p 29-38. Die Olefinmetathese–neue Katalysatoren vergr??ern das Anwendungspotential, M. Schuster and S.Blechert, Chemie in unserer Zeit, 1, 24-29, 2001. Classics in Total Synthesis II, K.C. Nicolaou and S.A. Snyder, Wiley-VCH, Weinheim, 2003.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号