“消相反应”在合成中的应用
- 2012-07-09
- 专题
对于比较剧烈的放热反应,一般通过低温下缓慢滴加反应物来进行调控。这种反应条件一方面使操作变得复杂,另一方面需要额外消耗能量来维持低温,这对于大规模工业化生产是非常不利的。近年来,随着氟化学的发展而诞生的“消相反应”(Phase-Vanishing Reactions)[1]很好地解决了这一问题。利用“消相反应”,可使反应物和底物在反应初期被氟相间隔而不混合,并通过氟相相互进行微量传递,进而使得放热反应在室温下可控进行。这不仅大大简化了反应操作,而且无需额外消耗能量。从这点来看,“消相反应”在工业上具有潜在的应用价值。
这一领域的发展始于20世纪90年代初,Vogt 等[2]首先报道了氟溶剂和有机溶剂的相溶性具有温度依赖性。接着Horvath等[3]发明了氟两相催化(Fluorous Biphasic Catalysis, FBC) 体系来回收催化剂。这一新概念的提出,立即引起了化学界的广泛关注。在1996 年后,氟化学在有机合成领域的应用得到了飞速发展,一些重要的进展包括氟相分离(Fluorous Separations) 技术[4~10]、氟相组合化学(Fluorous Combination Chemistry)[11]、氟三相反应(Fluorous Triphasic Reactions)[12]、“消相反应”(Phase-Vanishing Reactions)[1]等。本文主要介绍“消相反应”的研究进展,并简要介绍笔者在高分子合成方面的初步研究工作,其它方面的研究进展可参见近期的综述[13~22]。
1 “消相反应”的原理
1.1 基本原理
“消相反应”是氟三相反应的一种变体,其机理有两种[23]。一是扩散机理,第一相的有机或无机底物扩散到氟相(第二相),然后在第三相与底物反应。在反应过程中,反应物相消失,由此而命名为“消相反应”。另一是萃取机理,先通过溶剂扩散,顶部相密度逐渐增大,底部相密度逐渐减少;当顶部相密度接于中间氟相密度时,就以小液滴的形式穿过氟相到达底物相;小液滴中溶有部分反应物,这样就使得反应物和底物相接触并发生反应。扩散机理限于能溶于氟相的底物或反应物,而萃取机理适用于不溶于氟相的反应物。
1.2 基本过程
在“消相反应”中,多使用氟溶剂作为间隔介质,使有机底物和反应物在反应初期不相互混合。这主要是利用氟溶剂特殊的物理性能,即氟溶剂和大多数有机试剂不相溶,而且比大多数有机试剂重[12]。如果反应物(如卤化物)密度比氟溶剂大,则反应可在试管中进行(图1)。反应结束后,产物留在上层有机相而底部则出现相消失。如果底物和反应物都比氟溶剂轻,则相消失反应可在U 形管中进行(图1)。当反应结束时,底物和反应物相中必有一相消失[22]。
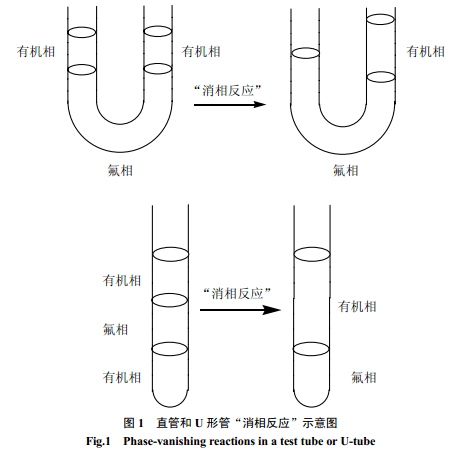
2 “消相反应”的应用
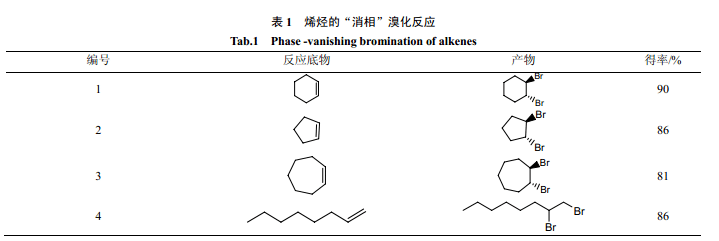
2.1 溴化反应
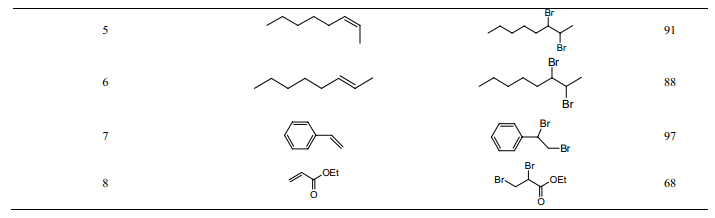
2002 年,Curran 等[12]最先报道了“消相反应”。他们在室温下,以全氟己烷为屏蔽相,用液溴对不同的烯烃进行溴化反应(表1)。当反应进行完全时,液溴层消失,氟相恢复透明。研究表明,在保持三相体系条件下,对液溴层进行缓慢的搅拌能大大提高反应速度。
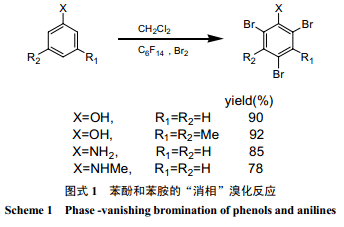
Verkade等[24]利用“消相反应”,用液溴对酚类和苯胺类有机物进行了溴化,得率较高(图式 1) 。
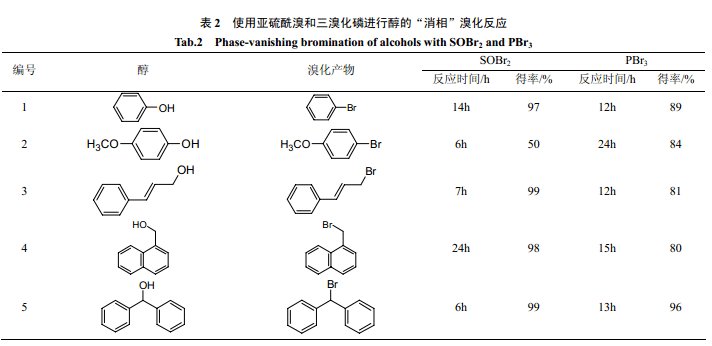
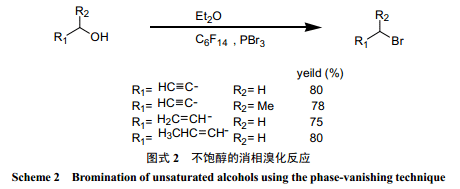
Ryu 等[25]分别用亚硫酰溴和三溴化磷进行了醇的消相溴化反应。在消相反应中,亚硫酰溴的溴化效果明显好于三溴化磷。另外从反应的得率来看,也比较高。由于消相反应存在传质过程,所以反应时间要比单一液相反应长。Verkade等[24]用三溴化磷对带炔丙基和烯丙基的不饱和醇进行了消相溴化反应(图式 2)。
2.2 氯化反应
Jernej 等[26]在氟三相体系下,用氯气对烯烃进行氯化反应。这个反应利用了氟溶剂易溶解氯气的特性。当烯烃被完全消耗后,底物相由于溶解了氯气而变黄,所以通过有机相颜色的变化可以判断底物是否反应完全。和许多用氯气直接氯化的反应相比,在U 形管中通过氟相传递的氯化反应有许多优点:第一,反应相和本体反应物是分离的,这点对于像氯气这样非常活泼、危险和强腐蚀性的物质来说尤其重要;第二,通过氟相传递氯气的过程是比较缓慢的,这种缓慢加入反应物的过程对于活泼的反应物和放热反应非常有利;第三,氟溶剂的回收利用率高。

Ryu 等[25]在U 形管装置中,分别用亚硫酰氯和三氯化磷进行了醇的氯化反应。他们还用亚硫酰氯进行了平行氯化反应,验证了三相U 型反应体系也适用于平行反应。
2.3 氧化反应
Verkade等[24]用间氯过氧苯甲酸(m-CPBA )进行了胺、硫化物和烯烃的“消相”氧化反应,得率接近文献报道的最大值。在三相体系中,m-CPBA 溶于密度大于全氟己烷的1,2-二溴乙烷,形成下层有机相;底物溶于一般的有机溶剂,形成上层有机相;氟相作为中间间隔介质。完全反应时,上层或下层有机相消失。后来Curran 指出Verkade进行的“消相”氧化反应机理属于萃取机理。上层或下层有机相消失取决于最终有机相的密度。Vrkade使用1,2-二溴乙烷来溶解比氟溶剂轻且难溶于氟相的有机物,这一方法大大拓宽了“消相反应”的应用范围。

2.4 烷基化反应和脱烷基化反应
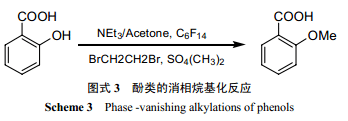
利用硫酸二甲酯进行酚类的烷基化反应一般要在回流条件下进行。但 Verkade等[24]发现,利用“消相反应”,在室温下就可用硫酸二甲酯进行水杨酸的烷基化反应,得率高于以往文献报道的最大值(图式 3) 。
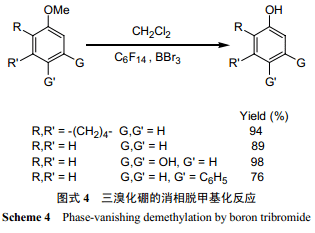
三溴化硼是高效的醚类脱烷基化试剂,而且其密度比全氟己烷大。三溴化硼的脱烷基化反应是个高度放热反应,一般需要在低温下进行。但 Curran 等[1]利用“消相反应”,在室温下用三溴化硼对苯甲醚进行了脱甲基化反应(图式 4)。
2.5 付氏酰基化反应
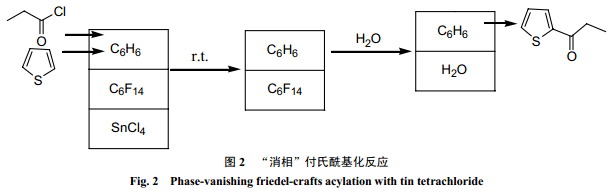
Ryu 等[27]利用消相反应,成功地用四氯化锡对噻吩进行了付氏酰基化反应。在三相体系中,噻吩和丙酰氯溶于苯形成上层有机相,四氯化锡形成底相,全氟己烷作为中间间隔介质。反应结束时,底相消失,产物从上层有机相得到(图2) 。
3 氟三相体系下甲基丙烯酸甲酯的氧化还原自由基聚合
氟三相体系以及消相反应的应用目前主要集中在有机物合成以及产物分离方面,在高分子合成研究领域的应用尚鲜见报道。笔者在氟三相体系下进行了聚合反应尝试,并成功地实现了甲基丙烯酸甲酯氧化还原自由基聚合,在引发剂侧得到了分子量分布较窄的聚甲基丙烯酸甲酯,多分散系数为1.38 ,反应示意图如图 3 所示[28]。

实验结果表明,氟试剂的屏蔽作用使得引发剂和单体相分离,导致两侧自由基浓度产生巨大差异,并最终使得两侧所得聚合物的分子量和多分散系数明显不同。引发剂侧之所以能得到分子量分布较窄的聚合物,很可能是由于该侧自由基浓度大而单体浓度低所致。目前,该聚合体系的动力学正在进一步研究之中,有望产生一种新的高分子合成方法。
4 结语
虽然消相反应体系的研究仍处于初始阶段,但是许多研究成果表明消相反应在合成上有着巨大的应用潜力。利用消相反应,在室温下可使放热反应缓慢和可控地进行,无需外加设备。消相反应的节能优点,使它具备了大规模工业化生产的潜力。但氟试剂价钱高、环境污染和对人体有害等是该技术的不利因素。反应时间长是该技术的另一个明显不足。因此,目前该技术的一个重要研究方向是通过调节氟三相体系的溶剂组成、用量和相界面的截面积来提高有机物在氟相的扩散速度,缩短反应时间[23]。相信随着氟相技术的不断发展,其工业化进程必将大大加快。
参考文献
[1] I Ryu, H Matsubara, D P Curran et al. J. Am. Chem. Soc., 2002, 124: 12946~12947.
[2] D W Zhu. Synthesis, 1993: 953~954.
[3] I THorvath, T Rabai. Science, 1994, 266: 72~75.
[4] D P Curran. Synlett, 2001: 1488~1496.
[5] D P Curran, Z Luo. J. Am. Chem. Soc., 1999, 121: 9069~9072.
[6] D P Curran, Y Oderaotoshi. Tetrahedron, 2001, 57: 5243~5253.
[7] W Zhang, Z Luo, D P Curran. J. Am. Chem. Soc., 2002, 124: 10443~10450.
[8] Y Nakamura, S Takeuchi, K Okumura. Tetrahedron Lett., 2003, 44: 6221~6225.
[9] H Matsuzawa, K Mikami. Tetrahed Lett., 2003, 44: 6227~6230.
[10] H Matsuzawa, K Mikami. Synlett., 2002: 1607~1613.
[11] Z Luo, Q Zhang, D PCurran. Science, 2001, 291: 1766~1769.
[12] N Hiroyuki, L Bruno, D P Curran et al. J. Am. Chem. Soc., 2001, 123: 10119~10120.
[13] I T Horvath. Acc. Chem. Res., 1998, 31: 641~650.
[14] E Wolf , G Koten, B Deelman. J. Chem. Soc. Rev., 1999, 28: 37~41.
[15] M Cavazzinni, F Montannari, G Pozzi et al. J. Fluorine Chem., 1999, 94: 183~193.
[16] D P Curran. Med. Res. Rev., 1999, 19: 432~438.
[17] D P Curran. Pure Appl. Chem., 2000, 72:1649~1653.
[18] J A Gladysz, D P Curran. Tetrahedron, 2002, 58: 3823~3825.
[19] C C Tzschucke, C Markert, W Bannwarth et al. Angew. Chem. Int. Ed., 2002, 41: 3964~4000.
[20] A P Dobbs, M R Kimberley. J. Fluorine Chem., 2002, 118: 3~7.
[21] J I Yoshida, K Itami. Chem. Rev. 2002, 102: 3693~3716.
[22] W Zhang. Chem. Rev., 2004, 104: 2531~2556.
[23] D P Curran. Org. Lett., 2004, 6: 1021~1204.
[24] N K Jana, J G Verkade. Org. Lett., 2003, 5: 3787~3790.
[25] H Nakamura, T Usui, I Ryu et al. Org. Lett., 2003 , 5 :1167~1169.
[26] I Jernej, S Stojan, Z Marko. Chem. Commun., 2003, 2496~2497.
[27] H Matsubasa, S Yasuda, I Ryu. Synlett, 2003: 247~249.
[28] S Z Chen, Y P Bai, Z L Li. Chinese Chemical Letters, 2006, 17:258~260.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号