高效毛细管电泳法在酶反应动力学研究中的应用
- 2012-07-11
- 专题
1967年Hjerten[1]最早提出毛细管电泳技术,并在3mm内径的管子中使用高场强进行自由溶液的区带电泳(CZE) 。到了20世纪80年代,毛细管电泳技术取得了突破性的进展,Jorgenson等[2]用75μm 的毛细管,采用柱上荧光检测成功分离了氨基酸及多肽物质,获得了40万的理论塔板数,并实现了正、负离子的同时分离。近几年来毛细管电泳技术已经成为分析化学中发展最为迅速的领域之一,它具有快速、高分辨率、高灵敏度和样品用量少、成本低等优点,在生物分析和生命科学领域中有极为广阔的应用前景。
1 毛细管电泳在酶反应动力学中的应用
酶反应的基本动力学关系,是指酶反应速度与酶和底物间的动力学关系。描述这种基本动力学关系的是米氏方程(Michaelis-Menten equation) 。该方程提供了两个极为重要的酶反应动力学常数:米氏常数Km 和转化率Kcat,通过这些参数表达了酶反应性质、反应条件和酶反应速度的关系。其中,Km 是一个特征常数,Km 越大,说明酶和底物的亲和力越小,越容易解离。所以,在酶反应动力学中,关键是要测得其米氏常数Km。
测定酶反应动力学的方法很多,主要包括分光光度法、荧光法、放射免疫法以及薄层和液相色谱法等。近年来,毛细管电泳法以其进样量少、操作简便、易于分离和定量、无需引入有机溶剂等优点,越来越广泛地应用于酶体系的测定中。毛细管电泳不但具有多种分离模式(如区带毛细管电泳CZE 、胶束电动毛细管电泳 MEKC 、凝胶毛细管电泳 CGE 、亲和毛细管电泳 ACE 、等电聚焦毛细管电泳CIEF、毛细管电色谱 CEC 等),还有多种检测模式(如紫外、二极管阵列 PDA 、荧光[3]、电化学检测器等),更加适于酶反应动力学的测定。
在毛细管电泳法测定酶反应动力学参数的研究中,最常用的测定酶活力的方法是区带毛细管电泳法[4]。Zhang 等[5]用区带毛细管电泳法测定了血管紧张素转化酶(ACE) 的活性、米氏常数和最大反应速度。在式(1)所示酶反应体系中,以产物峰面积对浓度进行回归分析,得到线性回归方程,通过监测不同底物浓度下产物的峰面积,计算出酶活力,根据米氏方程用Lineweave-Burk作图法得到米氏常数和最大反应速度。此法分离重现性高,分析速度快,试剂消耗量少。酶与底物总量仅需要150μL,并且无需加入有机溶剂,6min即可完成分离,8 次测定迁移时间及峰面积的相对标准偏差可分别达到0.15% 和0.71% 。

区带毛细管电泳法易于控制,但是有些酶反应底物和产物用区带毛细管电泳法很难达到分离的目的,所以也有不少研究者[6~8]利用胶束电动毛细管色谱法进行分离,被分离组分根据本身疏水性的不同达到分离,疏水性越强,作用力就越大,迁移时间就越长。Choi 等[9]就是采用MEKC分离法,基于胶束和水相的分离,研究测定分泌腺磷酸脂酶的活性。此法能够准确定量底物和产物,测得值经放射性测定法比较,结果可靠。Yoshimoto等[10]用亲和毛细管电泳法进行酶动力学常数的测定,通过桥基基团共价作用将酶键合在石英毛细管内壁上,然后引入底物,产物与未反应的底物在电渗流的作用下到达检测端,由产物的峰高进行定量分析,由Lineweaver-Burk作图法得到米氏常数,此法节省试剂,仅需要μg级的酶和ng级的底物就可以用来分析测试。
胶束电动毛细管电泳法可以使分子结构相似、分子量相近的底物和产物得到基线分离,但是在酶动力学反应体系中要慎重选用表面活性剂。胶束法最常用的是阴离子表面活性剂SDS ,但SDS 是蛋白质的变性剂,因此选用SDS 测定酶反应动力学参数时,只能适用于柱外反应;有些疏水酶在分离过程中不出峰或者因吸附产生的峰严重拖尾,这种情况下可以在缓冲溶液中加入阳离子表面活性剂(如CTAB等)或适量的有机溶剂(如乙腈等),但应注意阳离子表面活性剂极易使电渗反向,检测时注意加反向电压;有些非离子表面活性剂(如BRIJ-35) 也可以用于酶的分离与检测。
在毛细管电泳研究酶反应动力学的过程中,进样方法是值得探讨的问题之一。多数酶反应采用柱外反应(off-line mode),但是柱外反应浪费试剂,所以近年来柱上反应模式(in-line mode) 发展迅速,尤其对于相对不太稳定的物质,用柱上反应法有其特别的优势[11]。在柱上酶反应体系中,电泳媒介微分析(EMMA) 法[12]是最常用的进样方法,基于各组分电泳淌度的不同实现柱上反应并得到分离。EMMA法主要有两种模式:连续模式(continuous mode or long contact mode)和区带模式(plug-plug mode or transient format or short contact mode) 。连续模式是将一种反应物溶解于背景电解质并填充在毛细管中,另一种反应物以进样的形式引入到毛细管,两种反应物在毛细管内充分反应,加电压实现分离,这种模式比较浪费试剂,所以应用相对较少;区带模式应用于酶反应主要有三种类型:经典区带模式(classical plug-plug mode) 、部分填充技术(partial filling technique)和进样端反应模式(At-inlet reaction)。
有报道[13]采用电泳媒介微分析法(classical plug-plug mode) 与部分填充毛细管技术相结合,可达到反应和分离的目的。电泳媒介微分析法是将底物和酶注入毛细管,淌度较小的先进样,加一段低电压,由于电泳淌度不同,两区带相遇直至完全重叠,充分反应后,加高电压实现分离。部分填充技术是指部分毛细管填充酶反应最适缓冲溶液,其它部分填充背景电解质,用于底物和产物的分离。两种方法结合使得分离快速,易于自动化。
Sigrid 等[14]在研究血管紧张素转化酶的在线动力学反应中,采用了一种新的三明治式进样模式(At-inlet reaction),如图1所示。控制毛细管温度 37℃,进样端采用水-酶-底物- 酶-水的夹心模式,等待底物(马尿酰组氨酰亮氨酸HHL)和血管紧张素转化酶(ACE) 反应一段时间后,再通入2-[4-(2-羟乙基)-1-哌嗪]乙基磺酸HEPES缓冲溶液进行分离,在230nm 下通过检测不同底物浓度下产物的峰面积,通过米氏方程,由 Lineweaver-Burk作图法,得到 1/ v对1/[s]的双倒数曲线( 其中v为反应速率,[s]为底物浓度),曲线与横轴的交点即为-1/K m,由此得到米氏常数值。另外,在不影响重现性及分离度的情况下,还可以实现从短端进样,如在测定PDE 的动力学参数实验中[15],可以大大缩短分离时间,提高分析速度。
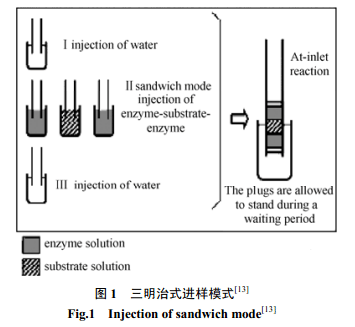
虽然EMMA法较柱外反应法节省试剂,使反应物的用量由μL 变为nL数量级,易控温,操作简便,省时,但是由于毛细管内径较细,柱内反应不够彻底,也不好控制。夹心式进样法由于扩散的影响,反应迁移时间的重现性较差;而经典的EMMA法,由于加两段相差很大的电压,使得基线不平,检测灵敏度降低。这些都是需要进一步改进的。
另外,在酶反应动力学的研究中,万谦等[16]采用多次进样技术测定蛋白激酶A 活力,发展了连续进样技术,使能在一个电泳过程中分析十个以上的样品,通过在线检测进行积分定量,大大节省了分析时间及费用。
表1 总结了近三年来毛细管电泳技术在酶反应动力学参数测定方面的应用。

2 毛细管电泳在抑制动力学研究中的应用
抑制剂是对酶反应速度有十分重要影响的一种因素,体外测定某些物质对酶反应 K m 的影响,往往有助于推断该物质可能有的生理效应,同时也可以帮助人们了解酶在机体内可能有的调节功能。抑制是指抑制剂与酶的活性有关部位结合后,改变了酶活性中心的结构(构象) 与性质,从而引起酶活性下降的一种效应。由酶抑制反应动力学可以判断抑制类型、计算抑制率和半数抑制浓度(IC50)。对于酶反应抑制剂的筛选,可用于研发新药,对于药疗卫生事业具有很大的意义。
在高效毛细管电泳测定酶抑制剂活性的研究中,一般是基于已经建立的酶反应动力学方法,
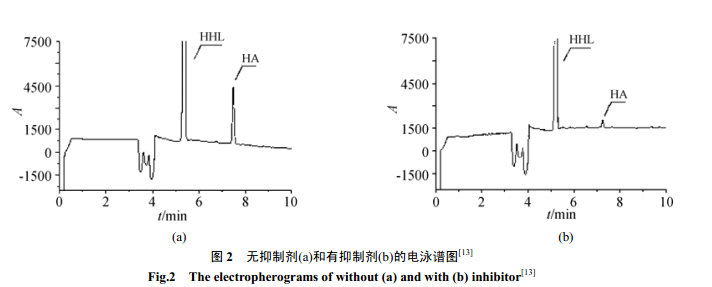
在底物中添加不同浓度或不同类型的抑制剂,同时作对照实验,根据抑制反应前后峰面积的变化计算抑制率,由抑制率对抑制剂浓度作图可以得到半数抑制浓度(抑制50%酶活性所需的抑制剂浓度),用来衡量抑制剂对酶的抑制效果,进一步通过抑制反应动力学曲线判断该反应的抑制类型。辛等[24]基于此法建立了高效毛细管电泳法测定血管紧张素转化酶抑制剂 Captopril 的活性的方法,经过酶反应动力学判断Captopril 为竞争性抑制剂,该法简便快速,7min内即可完成分离,除分析Captopril 对ACE 的抑制活性外,还可分析其它抑制剂对ACE 的抑制活性,并有可能用于筛选新的同类型药物。Sigrid 等[25]研究了在线抑制动力学检测法测定ACE 的五种抑制剂,用三明治的进样模式,柱上反应一段时间后,在230nm下进行分离检测,结果表明,加入抑制剂后,产物的峰面积比对照组有明显的减少,如图 2 所示,抑制效果明显,由此可通过公式(2)计算抑制率。
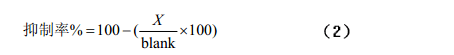
式中X 为加入抑制剂后产物的峰面积;blank为对照组产物的峰面积。
Chang 等[26]建立了用毛细管内微萃取技术测定ACE 抑制剂的活性,复杂样品无需预处理,4min内即可完成测定,此法快速、选择性好,适于筛分酶的有效抑制剂。
抑制剂大体可以分为两种类型——可逆抑制剂和不可逆抑制剂,而根据可逆抑制剂、底物和酶三者的相互关系,可逆抑制剂又可分为竞争性抑制剂、非竞争性抑制剂和反竞争性抑制剂。根据抑制反应动力学及米氏方程可以判断抑制类型。Angela等[27]研究了毛细管区带电泳法测定碱性磷酸酯酶的可逆抑制剂、不可逆抑制剂及其活性,采用激光诱导荧光检测器,酶的抑制作用通过产物所产生的荧光信号的强弱来测定。
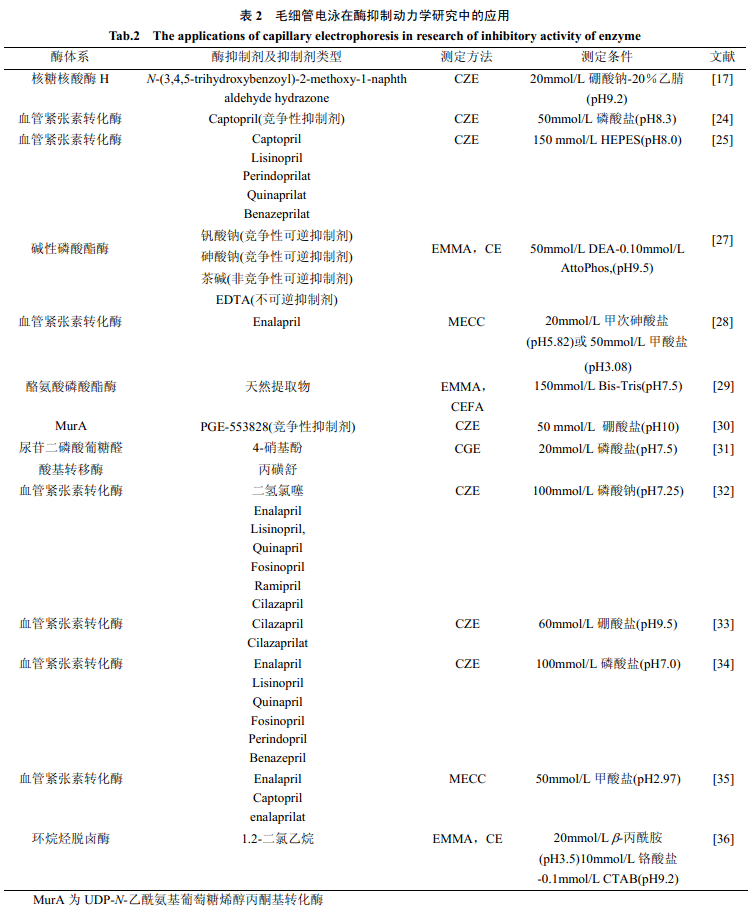
表2 是近年来毛细管电泳技术在酶抑制动力学研究方面的一些进展。
3 毛细管电泳(CE)在蛋白质-配体络合常数测定中的应用
酶与底物,酶与抑制剂的相互作用关系以及相互作用的位点是酶反应动力学必然要探讨的问题,所以有必要研究毛细管电泳法在蛋白质-药物、蛋白质-蛋白质络合常数测定中的应用。在毛细管电泳法测定蛋白质络合常数的方法中,主要分为两类:平衡CE法(equilibrium method)和非平衡CE法(nonequilibrium method)。
3.1 平衡CE法
平衡CE法是将一种络合组分加入到分离缓冲液中,另一络合组分以进样方式进入缓冲液,二者达络合平衡后进行分离,通过迁移时间的改变或者荧光光谱性质的改变来监测蛋白质络合的相互作用。平衡CE法主要适用于快速络合体系,大体有以下三种模式[37,38]:
3.1.1 HD 法(Hummel and Dreyer method) HD 法[39]是将一种物质(蛋白质或配体)加入到缓冲溶液中,另一种物质以少量进样的方式通过毛细管,两种物质在毛细管中达到络合平衡。如果蛋白质-配体络合物以及自由蛋白质的迁移速度近似相等,并且二者都比未被络合的配体的迁移速度快,那么蛋白质以及蛋白质-配体络合物将会和未被络合的配体相分离,如图3 所示。由于样品中总的配体浓度与缓冲溶液中的配体浓度相同,而络合物的形成和迁移会引起自由配体浓度的改变。因此可以检测到两组峰:正峰对应已络合的配体浓度,负峰对应自由配体浓度。HD法通过分析物峰面积与浓度成线性关系来进行分析测定,可以测得络合常数。

HD法广泛应用于可溶性配体和蛋白质络合常数的测定,尤其对于检测具有对映体结构的药物,灵敏度很高。HD法的缺点是自由蛋白和络合物两峰难于分离,解决办法可以通过修饰电荷来改变二者的淌度,或者对蛋白质进行化学衍生,采用荧光检测,尽可能实现分离。
3.1.2 ACE法(Affinity capillary electrophoresis) ACE 法测定络合常数的基本原理是基于各物质之间的淌度不同而得到分离的。ACE 法同HD 法相似,也是将一种物质加入到缓冲溶液中,另一种物质以进样的方式通过毛细管,两种物质在毛细管中达到络合平衡。与 HD法不同的是,ACE法是通过未络合和已络合分析物的电泳淌度的变化来计算络合常数。
此法适合研究大阴离子或游离目标蛋白质和配合物间电泳淌度有明显差别的物质。在蛋白质-DNA ,带电荷多聚物 -多肽相互作用的研究中应用广泛。但是 ACE 法不适宜分析高度亲和体系,特别是抗原-抗体之间的相互作用[41]。
3.1.3 FA 法(Frontal analysis method) FA 法[42~44]是在毛细管中填充缓冲溶液,再将大量样品(包括相互作用的各个物质)注入到毛细管中。如图4所示,迁移过程中蛋白质和蛋白质-配体的络合物由于迁移速度快先到达检测端,而自由配体分子后到达检测端,因此检测端出现两组峰,通常以自由配体浓度峰的峰高进行定量,可测络合常数值在103~108范围内。
FA法的缺点在于进样量大、试剂消耗多,而且检出限相对较高。FA法特别适合于高度键合的疏水性药物络合常数的研究测定。
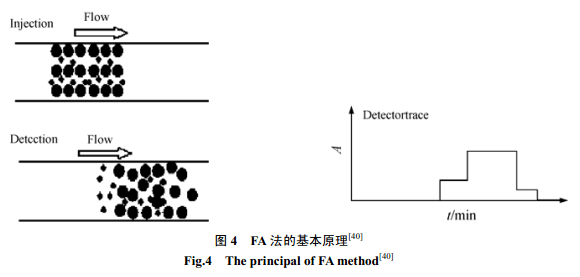
目前研究比较多的是利用ACE 法测定蛋白质的络合常数[45~47]。Varenne等[48]用ACE法研究了抗凝血酶与岩藻低聚糖的络合性质,将不同浓度的多糖注入到背景电解质溶液中,然后采用中性标记物-分离电解质-酶-分离电解质的进样方式,使酶和电解质中的多糖在毛细管中实现快速的络合- 解离动力学过程,最终通过有效淌度的计算得到络合常数。由于蛋白质和配体的快速的络合- 解离的动力学过程,导致自由蛋白质和其络合形式的绝对淌度有所差别。蛋白质的峰位置随着背景电解质中配体浓度的不同而改变,因此,可以通过蛋白质有效淌度的计算求得络合常数Kf。络合常数的计算与具体体系相关,计算公式也随体系的不同而不同。
3.2 非平衡CE法
非平衡CE法是将所有络合组分预混和,待平衡后,通过电泳使自由组分和络合组分得到分离。非平衡CE法只能用于解离速率相对较慢的络合反应,因为解离速率快的反应分离时间过长,导致检测不到络合物。随着 CE分离技术的不断发展,加上电泳数据分析数学模型的建立,非平衡CE法已经越来越多的应用于动力学研究中[49,50]。非平衡CE法包括NECEEM(nonequilibrium CE of equilibrium mixtures)模式和APCE(affinity probe CE)模式。NECEEM模式灵敏度高,可用于合成核苷亲和探针的灵敏度分析[51]。APCE 法理论上可以分离多种蛋白质形式,但是它的条件要求比较苛刻,可行性较差。
Okhonin等[52]用NECEEM法结合SweepCE 法测定了蛋白质-DNA 络合物二元分子速率常数,使用荧光检测体系。此法不要求蛋白质和 DNA形成络合物时荧光光谱发生变化,只要求蛋白质和DNA的淌度不同即可测定。
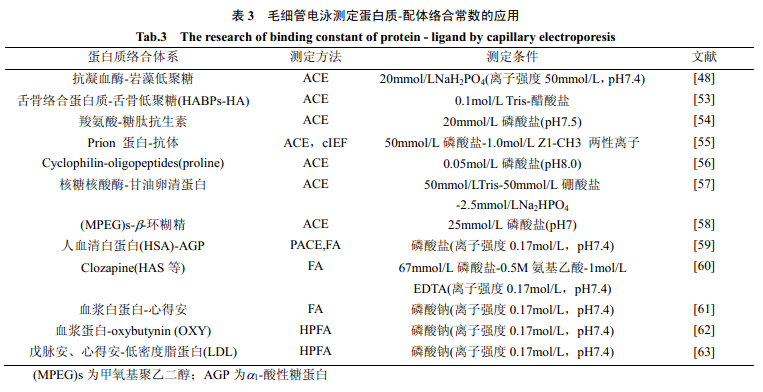
表3总结了近年来毛细管电泳技术在测定蛋白质-配体络合常数方面的应用。
4 结语
毛细管电泳法在研究酶反应动力学方面已经取得了相当大的进展,它具有分析速度快、试剂消耗少等优点,但仍然存在一些问题。比如毛细管电泳法最大的缺点就是重现性较差,在酶反应动力学研究中,很多文献报道的相对标准偏差均大于7%,影响了动力学研究的准确性,是迫切需要改进的。 随着毛细管电泳技术的不断发展,一些问题(如灵敏度低,重现性差等)将会逐渐得到解决,该技术在酶反应动力学研究中的应用也将会越来越成熟。特别是多元毛细管电泳,芯片毛细管电泳(MC-CE) 联用[64]等新技术的提出,更有利于拓展和深入研究各种复杂酶的动力学关系及其应用。
参考文献
[1] S Hjerten. Chromatogr. Rew., 1967, 9: 122~134.
[2] J W Jorgenson, K D Lukacs. Anal. Chem., 1981, 153: 1298~1302.
[3] M Berezovski, W P Li, P C Dale et al. Electrophoresis, 2002, 23(19): 3398~3403.
[4] J R Anderson, O Cherniavskaya, I Gitlin et al. Anal. Chem., 2002, 74(8): 1870~1878.
[5] R Z Zhang, X H Xu, T B Chen. Anal. Biochem., 2000, 280: 286~290.
[6] V D Sigrid, A V Schepdael, J Hoogmartens. Electrophoresis, 2002, 23: 2854~2859.
[7] M Kulp, M Kaljurand. J. Chromatogr. A, 2004, 1032: 305~312.
[8] M Kulp, M Kaljurand, T Kaambre et al. Electrophoresis, 2004, 25(17): 2996~3002.
[9] S Choi,Y S Lee, D S Na et al. J. Chromatogr. A, 1999, 853: 285~293.
[10] Y Yoshimoto, A Shibukawa, H Sasagawa et al. J. Pharm. & Biomed. Anal., 1995, 13(4/5): 483~488.
[11] N Soň a,V D Sigrid,G Zdenek et al. J. Chromatogr. A, 2004, 1032(1-2): 319~326.
[12] N Soň a, V D Sigrid,V Schepdael et al. J. Chromatogr. A, 2004, 1032(1-2): 173~184.
[13] N Soň a, Z Glatz. Electrophoresis, 2002, 23: 1063~1069.
[14] V D Sigrid, S Vissers, A V Schepdael et al. J. Chromatogr. A, 2003, 986: 303~311.
[15] X Cahours, C Viron, Ph Morin et al. Anal. Chimica Acta, 2001, 441: 15~21.
[16] 万谦,陈长征,屠红 等. 生物化学与生物物理学报, 1999 ,27(5):579~584.
[17] K C Chan, S R Budihas, S F J L Grice et al. Anal. Biochem., 2004, 331: 296~302.
[18] E S Dustin, Y Abdelaziez, C H Ahn et al. Anal. Biochem., 2003, 316: 181~191.
[19] Y Kanie, O Kanie. Carbohydrate Research, 2002, 337: 1757~1762.
[20] J Jitkova, C N. Carrigan, C D Poulter et al. Anal. Chimica Acta, 2004, 521: 1~7.
[21] Y Murakami, T Morita, T Kanekiyo et al. Biosen. & Bioelectron., 2001, 16: 1009~1014.
[22] H W Xu, G E Andrew. Anal. Bioanal. Chem., 2004, 378: 1710~1715.
[23] T L Paxon, T S Brown, H N Lin et al. Anal. Chem., 2004, 76: 6921~6927.
[24] 辛志宏,马海乐,吴守一 等. 药学学报, 2003, 38(11): 843~843.
[25] V D Sigrid, S Novakova, A V Schepdael et al. J. Chromatogr. A., 2003, 1013: 149~156.
[26] B W Chang, R L C Chen, I J huang et al. Anal. Biochem.,2001, 291: 84~88.
[27] A R Whisnant, S D Gilman. Anal. Biochem. ,2002, 307: 226~234.
[28] E Stellwagen, R Ledger. Anal. Biochem. ,2003, 321: 167~173.
[29] A Belenky, D Hughes, A Korneev et al. J. Chromatogr. A, 2004, 1053: 247~251.
[30] H J Dai, N P Christian, J J Bao. J. Chromatogr. B, 2001, 766: 123~132.
[31] S K Kumiko, K Masaru, T Toyo’oka. J. Chromatogr. A, 2004, 1051: 261~266.
[32] S Hillaert, K D Grauwe,W V Bossche et al. J. Chromatogr. A, 2001, 924: 439~449.
[33] J A Prieto, U Akesolo, R M Jimenez. J. Chromatogr. A, 2001, 916: 279~288.
[34] S Hillaert, W V Bossche. J. Pharm. & Biomed. Anal., 2001, 25: 775~783.
[35] E Stellwagen, R Ledger. Anal. Biochem., 2003, 321: 167~173.
[36] M Telnarova, S Vytiskova, M Monincova et al. Electrophoresis, 2004, 25:1028~1033.
[37] Y Tanaka, S Terabe. J. Chromatogr. B., 2002, 768: 81~92.
[38] M H A Busch, J C Kraak, H Poppe. J. Chromatogr. A, 1997, 777: 329~353.
[39] G Berger, G Girault. J. Chromatogr. B, 2003, 797: 51~61.
[40] R Freitag. J. Chromatogr. B, 1999, 722: 279~301.
[41] R M Seifar, R H Cool, W J Quax et al. Electrophoresis, 2004, 25: 1561~1568.
[42] K L Burns, S W May. J. Chromatogr. B, 2003, 797: 175~190.
[43] T Ohnishi, N A L Mohamed, A Shibukawa et al. J. Pharm & Biomedical Anal., 2002, 27: 607~614.
[44] Y Kuroda, Y Watanabe, A Shibukawa et al. J. Pharm & Biomedical Anal., 2003, 30: 1869~1877.
[45] J Kaddis, C Zurita, J Moran et al. Anal. Biochem., 2004, 327: 252~260.
[46] B Steinbock, P P Vichaikul, O Steinbock. J. Chromatogr. A, 2001,943:139~146.
[47] W B Zhang , L H Zhang , G C Ping et al. J. Chromatogr. B, 2002, 768: 211~214.
[48] A Varenne, P Gareil, S Colliec-Jouault et al. Anal. Biochem., 2003, 315: 152~159.
[49] M BereZovski, S N Krylov. J. Am. Chem. Soc., 2002, 124(46): 13674~13675.
[50] M BereZovski, S N Krylov. Anal. Chem., 2004, 76: 7114~7117.
[51] P L Yang, R J Whelan, E E Jameson et al. Anal. Chem., 2005, 77: 2482~2489.
[52] V Okhonin, M BereZovski, S N Krylov. J. Am. Chem. Soc., 2004,126(23):7166~7167.
[53] M Kinoshita, K Kakehi. J. Chromatogr. B, 2005, 816: 289~295.
[54] M Azad, C Silverio, Y Zhang et al. J. Chromatogr. A, 2004, 1027: 193~202.
[55] G D Li, X J Zhou, Y H Wang et al. J. Chromatogr. A, 2004, 1053: 253~262.
[56] S Kiessig, F Thunecke. J. Chromatogr. A, 2002, 982: 275~283.
[57] K Uegaki, A Taga, Y Akada et al. Anal. Biochem., 2002, 309: 269~278.
[58] C Karakasyan , M Taverna , M C Millot. J. Chromatogr. A, 2004, 1032: 159~164.
[59] Z J Jia, T Ramstad, M Zhong. J. Pharm. & Biomed. Anal., 2002, 30: 405~413.
[60] D W Zhou, F M Li. J. Pharm. & Biomed. Anal., 2004, 35: 879~885.
[61] T Ohnishi, N A L Mohamed , A Shibukawa et al. J. Pharm. & Biomed. Anal., 2002, 27: 607~614.
[62] A Shibukawa , N Ishizawa, T Kimura et al. J. Chromatogr. B, 2002, 768: 177~188.
[63] Y Kuroda, Y Watanabe, A Shibukawa et al. J. Pharm. & Biomed. Anal., 2003, 30: 1869~1877.
[64] A R Stettler, M A Schwarz. J. Chromatogr. A, 2005, 1063: 217~225.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号