窄带隙高分子光伏材料
- 2012-07-23
- 专题
由于能源紧张加上高分子光伏电池具有易加工、质量小、成本低等优点,各国越来越重视高分子太阳能电池的研究。高分子光电池之所以一直没有大规模实际运用,是因为光电转化效率还较低,迄今最高只有3.5%[1]。其主要原因是大多数活性材料最大吸收波段在350~650nm之间,而太阳光最大光子流在600~800nm,因此吸收光谱和太阳能不匹配;其次是光生载流子从活性材料传输到电极的损失,造成总能量效率一直不高。
研究窄带隙的高分子光伏材料和能吸光的受体材料可以增加对太阳光的利用率,在活性材料中具有电子和空隙各自传输通道可以克服载流子传输损失。本文主要回顾了近年来出现的窄带隙光伏材料,总结了物质带隙和分子链结构的关系,提出合成窄带隙光伏材料的一般思路;同时还对能够吸光的受体材料和能够形成载流子传输通道的“双缆”光伏材料进行了介绍。
1 窄带隙高分子光伏材料
尽管高分子光伏材料种类比较多,但由于性能和合成及加工方面的限制,实际应用并不多,仅限于不多的几种物质及其衍生物。以PPV 衍生物研究得最多,其中聚3-己基噻吩(P3HT)、MEH-PPV[56]、OC1C10-PPV(MDMO-PPV)等效果最为突出。但它们最大吸收波长在 500nm,不太令人满意。由于大多数活性材料最大吸收波段在350~650nm 之间,所以应该研究最大吸收波长在红光和近红外的具有窄能隙(<1.8eV)的共轭高分子。
1.1 高分子光伏材料带隙的影响因素
前人已经进行大量研究的高分子带隙 Eg和结构的关系主要有以下几点:(1)带隙和共轭链的交替链长有关,长度越短,带隙越小;醌式结构的分子链可以减少交替链长度,因此能够减少共轭高分子的带隙;(2)分子链中如果存在共轭的芳香单元,由于环间可能存在环扭转张力,因此影响共轭,不利于带隙的减少;(3)在有芳香单元的共轭链中,带隙还和芳香性有关。芳香性体现了环中π电子定域和离域的竞争,对局部芳香体系来说,芳香性越强,电子在芳香体系内离域越好。但对整个高分子分子链,如果芳香性太强,反而不利于π 电子在整个分子链中的离域;(4)带隙和分子链间作用力有一定关系。分子间相互作用越强,带隙越小。因此,通常物质在固体下比在液态下有更低的带隙,有序的相结构比无序的相结构有更低的带隙;(5)如果共轭链中存在大体积的单元或取代基,因空间位阻的影响会降低共轭效果,使共轭长度减少,导致带隙增加。总之,线性共轭高分子的带隙(Eg)可用下式表[2]
Eg=E(r) + E(θ)+ E(Res) + E(Sub) + E(Int)
式中各项表示各种影响因素对带隙的贡献值。E(r)与交替出现的单双键长度差有关;E(θ)由空间位阻等原因使得芳香环共轭体系不能很好地共平面所产生的扭转能决定;E(Res)是由芳香环的芳香共振能决定;E(Sub)代表了取代基对带隙的贡献;E(Int) 表示分子间相互作用的强弱对带隙的影响。虽然高分子光伏材料和普通导电高分子有所不同,但在考虑带隙和结构的关系时,基本原则是一致的。
1.2 给吸电子单元交替对带隙的影响
分子链由给电子单元和受电子单元交替构成时,可以使它们之间的单键的电子发生偏移,使之具有部分双键特征,从而减少交替键长,减小带隙。常用的吸电子单元有如 2,1,3-苯并噻二唑(B)、噻吩并[3,4-b]吡嗪、苯并[1,2-c:4,5-c']双[1,2,5]噻二唑、[1,2,5]噻二唑并[3,4,-g]喹喔啉;给电子单元有如双噻吩、N,N-二甲基双吡咯、2,5-二(2-噻吩基)- N -十二烷基吡咯(TPT)[3,4]。由于需要和受体混合,通常不能通过电化学聚合,因此寻找可溶的低带隙的 D-A 高分子非常困难,仅有少数物质[3]。
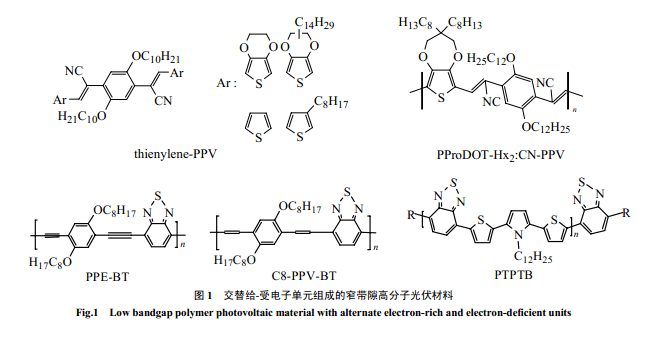
高分子能通过增加其最高占有轨道(HOMO)能量(减少氧化电位)或降低最低空轨道(LUMO)能量(减少还原电位)来减少带隙Eg,但是,前者会降低开路电压[3](分散异质结光伏电池 VOC主要是由给体的HOMO和受体的LUMO 能量差决定的[5]),后者则会减少在异质电池中激发态的电子转移到受体的驱动力;同时,太低带隙能够增强辐射衰退,影响激发态寿命和激子扩散长度[3]。因此,高分子光伏材料不同于本征导电高分子,并非带隙越低越好。Dhanabalan 等[3,6,16]考虑了这几方面的因素,选用了中强的给电子单元 TPT 和受电子单元B 合成了D-A交替构成的窄带隙的PTPTB(1.6eV),吸收可以延到750nm,构成的(PTPTB/PCBM)异质电池能量转换效率最高可以达到1%(AM=1.5)。路胜利等用给电子的 PPE单元[51]和PPV 单元[49]与2,1,3-苯并噻二唑共聚分别得低带隙的交替共聚物PPE-BT和C8-PPV-BT(1.77eV),吸收都分别比纯 PPE 和PPV 有较大的红移。
也可以通过引入取代基来改变单元的亲电性,在共轭链上分别交替引入给、受电子取代基同样可以减少带隙。2004年,Colladet等[7]合成了一系列 thienylene-PPV 衍生物窄带隙光伏高分子(1.55-1.94eV),由于它在 PPV 的苯撑上交替引入了受电子的氰基和给电子的烷氧基,所合成的高分子的分子量高且有较好的溶解性。Barry等[58]合成了PProDOT-Hx2:CN-PPV 共聚物,不仅带隙低(1.7eV),而且在空气中有很好的稳定性。
1.3 分子链上醌式结构对带隙的影响
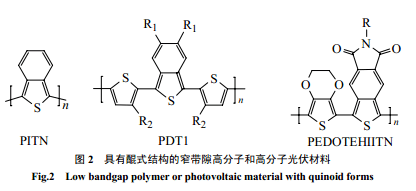
早在上世纪 80 年代中期,Wudl等[8,9]合成了窄带隙的导电高分子聚苯并噻吩(PITN) ,噻吩环和苯环稠联在一起,通过苯环来稳定醌式结构,从而使主链中醌式结构多于芳香结构。由于醌式结构有更低的带隙,因此降低了 PITN的带隙[10~12],双甲基、双甲氧基取代都不会影响此效果[12]。随后一系列 PITN 衍生物,包括含有噻吩的窄带隙材料相继合成,但醌式结构可使分子链具有刚性,即使引入侧链也不易溶解,不利于加工和应用[15],实际很少应用于光伏材料。为了解决此问题,一些能溶解的 PITN预聚体的合成[14,17],给 PITN衍生物用于光伏材料带来了希望。PDT衍生物聚(噻吩-苯并噻吩)和聚(1,3-双噻吩-苯并噻吩)[13,14]用于光伏材料取得了较好效果。Polec 等[18,20]用新的途径合成了被烷氧基等取代的单体双硫杂苯酞,通过热聚合合成了可溶的PITN衍生物聚(5,6-二硫杂辛基苯并噻吩),可以作为窄带隙光伏材料(1.2eV)。Cravino等[19]研究了带隙为1.1eV的PEDOTEHIITN(图2),它在太阳光最强光子流700nm处有很强的吸收,同时显示了优良的稳定性、溶解性和薄膜形成性能,是很有希望的光伏材料。
1.4 分子链间作用力对带隙的影响
分子链聚集态结构的有序化,可以增加分子链间的作用力,减少带隙。新型窄带隙光伏材料PEOPT可以通过一定的物理处理,使结晶度增加( 结构的有序化增加),带隙从2.1eV减少到1.75eV[22,23]。聚噻吩衍生物的局部规整减少了空间阻碍使噻吩环扭转较少,同时增加了分子链之间的作用,从而减少带[18]。苯环取代的分子链的聚集态结构具有一定的有序性,分子链间作用力增大,可以形成部分结晶结构,并且结晶程度还和苯环取代基上的柔性链取代位置(决定伸展方向)有关[24]。
拉电子的噻吩并[3,4-c][1,2,5]噻二唑衍生物(TTD)和2,1,3-苯并噻二唑(BTZ)单元的 S…N 作用力使得聚合物聚集态结构有序化,从而对降低带隙有利[2],因此在给受单元交替的光伏材料中引入此结构能进一步降低带隙,如果解决了溶解性问题,就有望在光伏材料中得到应用。

1.5 分子链中窄带隙单元对带隙的影响
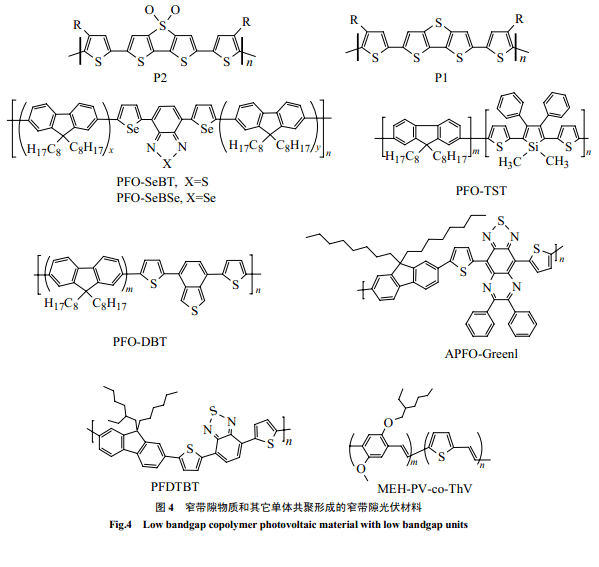
窄带隙导电高分子通常不熔不溶,带隙极窄,很难用于光伏材料。但可以通过和其它物质共聚来改性,以满足加工性、光电性等要求。聚芴是一种既有较高的空穴迁移率又比较稳定的宽带隙光电物质。它可以通过和其它窄带隙物质共聚,进行性质“剪裁”赋予共聚物以可能吸收红光、吸收波段增宽和溶解性好等特点。窄带隙物质单元通常由给-受体单元交替构成,噻吩由于具有较窄的带隙和较高的稳定性以及灵活的设计性能,一般作为给- 受体交替单元中的给电子体。Svensson 等[10]和周[25]分别合成了类似的新型光伏共聚物 PFDTBT 和PFO-DBT。它是由 9,9-二辛基芴和 7-双-2-噻吩基-2,1,3-苯并噻二唑共聚而得,吸收延展到红光,和PCBM 组成的异质结电池转换效率达到2%。芴与不同窄带隙的给-受-给单元的共聚物 APFO-Green1[15,64,65]和APFO-Green2[67]都具有吸收红移的特点,其中前者吸收更是达到 1000nm。Wan g等合成的PFO-TST[27]共聚物以及 PFO-SeBT(1.87eV)[26]和PFO-SeBSe[26,53](1.77eV) 共聚物,长波吸收增加,和 PCBM 组成的电池能量转化效率分别达到了2.01% 和1%(AM=1.5)。
PPV 良好的光电性能和成膜性能一直倍受关注,Hou等[57]用带隙较窄但成膜性能不好的聚 2,5-噻吩撑(PThV)和MEH-PPV共聚,得到的共聚物和 MEH-PPV比较,具有吸收红移、吸收波段增宽以及空穴迁移率增大的优点。Catellain等[28]通过共聚合成了P1、P2噻吩衍生共聚物,在窄带隙物质中引入烷基噻吩,使共聚物具有良好的溶解性,同时保持了窄带隙,P2甚至达到了1.7eV。
1.6 共轭芳香环间双键对带隙的影响
高分子光伏材料研究最多、性能较好的是 PPV 衍生物:MEH-PPV、OC1C10-PPV(MDMO-PPV)等。其中(MDMO-PPV/PCBM)总能量转化效率达到了迄今最高的 3.5%[1];这些物质主链由芳香环和双键交替构成,形成所谓的苯撑结构。由于存在双键,减少了环的空间阻碍,抑制了环的扭曲,有利于共平面,使共轭长度增加,带隙降低[11]。和PPV 相似,具有高分子量和高产量的聚噻吩撑(PTV)也具有此特点(1.55eV)[21]。现在设计低带隙芳环共轭高分子光伏材料时,多引入双键,以减少带隙。
1.7 取代基对光伏材料带隙的影响
共轭链上的取代基对光伏材料的性质有很大的影响,它有以下几个作用:(1)增加物质的溶解性。如果光电材料不溶,难于合成和成型,应用就有很大困难,没有实际应用价值。长链的线性烷基或烷氧基取代可以增加物质的可溶性。(2)增加分子链的有序性。一些取代基如苯环取代能增加取代基间或链间相互作用,可以提高链结构的规整性从而形成有序相,使带隙减少。但是长的烷基取代不利于规整性,同时在合成时要避免相邻取代基相撞,所谓形成头碰头(HH),尾碰尾(TT) 的缺陷从而造成空间位阻,影响分子链上邻近芳香环的共平面性,减小有效共轭。Roncali等[29]通过对聚 3-烷基噻吩的研究,指出线性烷基 C 原子 7~9 个共轭效果最好,但支链取代效果不好,可能是由于不利于分子间相互作用。(3)给、受电子取代基可以分别使共轭系统的 HOMO,LUMO 升高和降低,降低带隙。3,4- 乙烯基双氧噻吩(EDT) 比噻吩给电子强,它的衍生物共聚合体有很窄的带隙[31`33]和一定的溶解性[31],是很好的给电子体。
引入取代基不仅要考虑亲电性和增溶性,还要考虑共轭效应的影响[30]。侧链芳香环可以和主链形成共轭,增加共轭效果,有利于减少带隙。可用柔性烷基取代的苯基作主链的取代基,如PEOPT[22,23]。
当然,合成窄带隙光伏材料时,不应该考虑单一方面,各种影响因素都应该考虑。
尽管随着对窄带隙高分子结构的了解不断加深,有一些窄带隙的共轭高分子成功合成,但它们的外量子效率几乎都不高,很少有超过50% 的,这仍然影响光电效率的提高。同时,由于受到不溶不熔及低分子量的困挠,实际上不能应用于光伏材料[21]。因此合成具有良好溶解性,同时具有较高光电性能的高分子量的窄带隙物质仍然是一个挑战性的工作。
2 具有吸光性的受电子物质
受体物质大多数对可见光的吸收很小,如聚对-吡啶乙烯撑(PpyV)[45]、具有n 型半导体性质和低还原电位的含 N 杂环和主链有喹啉、嘧啶等结构的共轭高分子[60];部分二酰亚胺(如PPEI[63])以及TiO2[62]等无机粒子虽可以作为受体材料,但对可见光吸收很低。由于受体是活性层的主体部分,如常用的C60 衍生物 C60 PCBM 占75% ,极大制约效率的提高。
Wienk等[59]用吸光性增强、对称性不好的C70 PCBM 代替 C60 PCBM ,和 MDMO-PPV组成光电池,效率比 C60 PCBM 提高了50% 。高分子 BBL 能吸收红光(740nm) ,和给体组成的活性材料有较强的光捕获能力[46,47]。在共轭芳香环间的双键上或苯环上引入拉电子氰基可以提高对电子的亲和力,增加电子的迁移率,还可以提高阴离子的稳定性,因此 CN-PPV[48]可作吸光的受体。Helmut 等[50]合成了活性物质 Pc-C60(最大吸收在两个波段:<400nm,~700nm)可以作为分子给或受体光伏材料。但由于存在电荷传输障碍,构造的光电池转化效率并不好。此外,具有良好电子传导性的无机纳米半导体颗粒 CdSe也可以作为受体[61],它对长波可见光(720nm) 具有一定的吸收能力。

3 光伏材料中加入敏化剂增加光吸收
为了对太阳光进行“地毯”式的吸收,把窄带隙和宽带隙物质混合构成复合体,或把染料加入到共轭高分子中来增加对光的吸收。Winder[52]构造了(PTPTB/PCBM+Nile 红) 和(PTPTB/MDMO-PPV/PCBM)两种结构的光电池,发现前者效率有所提高,但后者却有所下降。对于前者,PTPTB 和Nile 都吸红光,贡献了光电流,作用机理是否是染料把激发电子转移给 PTPTB ,再通过 PTPTB 转移给受体,还是两者直接传输给 PCBM 还有待研究,但可以肯定染料形成的空穴的输运有困难。对于后一种模型光电池,发现PTPTB 对光电流的贡献远不及 MDMO-PPV。可以看出,这种粗糙的混合,性能并没有得到显著改善。但可以预料,随着对机理的不断理解,在此基础上进行改进,将是增加光利用率的一种有效途径。
4 双缆光伏材料
分子异质高分子光伏电池是把结构为给-受体的接枝或嵌段的共聚物作为光伏材料,通过一定的组装技术,给受体分别形成纳米级各自连续两相[34],构成分别传输电子和空穴的两种不同通道(图6,7) ,即所谓的“双缆”(双通道)高分子异质电池。通过精确控制给-受体嵌段长度,能够使柱的直径在激子的扩散距离内。因为激子容易扩散到界面分离,电子、空穴有连续的通道传输到各自电极,所以有较高的转换效率。目前已经报道的大体有以下几类。
(1)受体作为侧链连在给体高分子骨架上。侧链受体通常为C60 衍生物,主链为共轭的线性分子。2001 年Cravino等[35]把富勒烯吡咯烷通过用共价键和双噻吩相连,合成了具有光电属性的给体受体接枝单体。Ramosw 等[36]设计了(PPV 衍生物-C60)双缆异质光伏电池,获得了 Voc 为830mV ,Isc 为420μA/cm2(AM1.5) ,FF为0.29 ,EQE 为6%(480nm) 。近年来,这种结构的双缆材料研究较多[37~39]。
(2)给体-受体都作为侧链连在饱和脂肪链上。2004 年Lindner等[40]合成的嵌段共聚高分子具有骨架为饱和柔性σ 链,给体- 受体(杂环共轭体)以侧链形式和主链相连,同时受体嵌段具有吸光的特点。
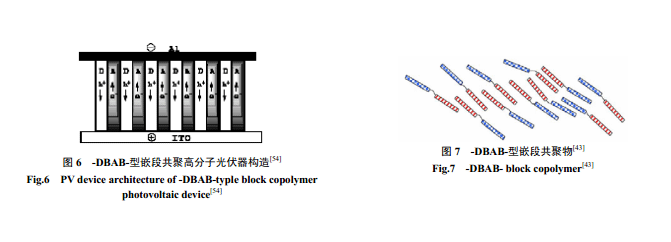
(3)-DBAB- 型线性嵌段共轭共聚物。2002 年Sun[43]设计了一种新的 DBAB 嵌段共轭共聚物(图6、图7) ,由于整个分子链都共轭,有利于电子传输。D是共轭给体嵌段,能吸收一定波长的光;A 是电子受体,也能吸收特定波长的光;B 是非共轭的绝缘柔性链(如脂肪单链),它能有效延缓电子、空穴的复合[44]。同时,σ键并不影响分子间和分子内的电子转移和激子的分离,这克服了-DA-型[41,42]电子和空穴复合太快太方便的缺点。此外,给-受体通过柔性链相连,还易于分子自组装形成柱状相分离。
5 高分子光伏材料发展方向和前景展望
为了提高太阳光的利用率和具有良好的载流子传输性能,高分子光伏材料可以从以下几个方面进行深入研究:(1)研究量子效率高、加工性能好的窄带隙光伏材料;(2)研究亲电性强、电子迁移率高、吸光性强的高分子电子受体;(3)弄清楚染料敏化等具有多种波段吸光性的材料复合体的载流子传输机理和本质,以便采取措施减少载流子的陷阱;(4)深入研究分子结构和聚集态结构对吸光性、吸光波段的关系以及对载流子的传输的影响,从而能够根据需要对光伏材料进行分子设计;(5)发展能够精确控制形态的“双缆”高分子光伏材料;(6)研究在氧气和水的条件下具有一定稳定性的光伏材料。
尽管高分子光伏材料已经取得了不少进展,但仍然有一些问题需要克服。高分子光电材料虽然很多,但真正能运用到光伏电池上却很有限,其中性能优良的窄带隙光伏材料更少,这极大制约了高分子光伏器件的发展和实际运用,因此开发新的高分子光伏材料特别是窄带隙材料成为当务之急。同时,一些高分子光伏材料外量子效率不高,合成路线不稳定可靠也不成熟,反应活性不高,预聚体稳定性或活性不高,合成的活性材料难溶难熔,不易加工,因此很难实际运用。 高分子光伏材料涉及到化学、光电子、固体物理等交叉学科,如果仅孤立从一个方面进行研究,是很难有突破的。但是,如今各种学科更多的研究者加入到该项研究中,对光伏材料的本质有了越来越深入的理解,因此高分子光伏材料的突破性进展很有希望。
参考文献
[1] F Padinger, R S Rittberger, N S Sariciftci. Adv. Funct. Mater., 2003, 13: 85~88.
[2] J Roncali. Chem. Rev., 1997, 97: 173~205.
[3] A Dhanabalan, J K J Van Duren, P A Van Hal et al. Adv. Funct. Mater., 2001, 11: 255~262.
[4] C Winder, N S Sariciftci. J. Mater. Chem., 2004, 14: 1077~1086.
[5] C J Brabec, A Cravino, D Meissner et al. Adv. Funct. Mater., 2001, 11: 374~380.
[6] J K J Van Duren, A Dhanabalan, P A Van Hal et al. Synth. Met., 2001, 121: 1587~1588.
[7] K Colladet, M Nicolas, L Goris et al. Thin Solid Films, 2004, 451-452: 7~11.
[8] M Kobayeshi, N Colaneri, M Boyse et al. J. Chem. Phys., 1985, 82: 5717~5723.
[9] J Wudl, M Kobayashi, A Heeger. Chem. Phys.,1984, 49: 3382~3384.
[10] J LJ bredas. Chem. Phys., 1985, 82: 3808~3811.
[11] C Quattrocchi, R Lazzaroni, J L Bredas et al. J. Phys. Chem., 1995, 99: 3932~3938.
[12] J L Bredas, A J Heeger , F Wudl. J. Chem. Phys., 1986, 85: 4673~4680.
[13] S E Shaheen, D Vangeneugden, R Kiebooms et al. Synth. Met., 2001, 121: 1583~1584.
[14] T S Hung, S A Chen. Polymer, 1999, 40: 3881~3884.
[15] X J Wang , E Perzon, J L Delgado et al. Appl. Phys. Lett., 2004, 85: 5081~5083.
[16] C J Braberc,C winder, N S Sariciftci et al. Adv. Funct. Mater., 2002, 12:709~712.
[17] H Meng, F Wudl. Macromolecules, 2001, 34: 1810~1816.
[18] I Polec, A Henckens,L Goris et al. J. Polym. Sci., Part A: Polym. Chem., 2003, 41: 1034~1045.
[19] A Cravino, M. A Loi, M C Scharber et al. Synth. Met., 2003, 137: 1435~1436.
[20] L Goris, M A Loi, H Neugebauer et al. Synth. Met., 2003, 138: 249~253.
[21] A Henckens, M Knipper, I Polec et al. Thin Solid Films, 2004, 451-452: 572~579.
[22] C J C Winder, M Scharber et al. Mater. Res. Symp. Proc., 2000, 598: BB3.24.1-BB3.24.6.
[23] C J Brabec, C Winder, M C Scharber et al. J. Chem. Phys., 2001, 115: 7235~7244.
[24] K E Aasmundtvei, E J Samuelsen, V V Mammo et al. Macromolecules, 2000, 33: 5481~5489.
[25] Q M Zhou, Q Hou, L P Zheng et al. Appl. Phys. Lett., 2004, 84: 1653~1655.
[26] R Q Yang, R Y Tian, J G Yan et al. Macromolecules, 2005, 38: 244~253.
[27] F Wang, J Luo, K Yang et al. Macromolecules, 2005, 38: 2253~2260.
[28] M Catellani, B Boselli, S Luzzati et al. Thin Solid Films, 2002, 403-404: 66~70.
[29] J Roncali, R Garreau, A Yassar et al. J. Phys. Chem., 1987, 91(27): 6706~6714.
[30] H A M Mullekom, J A J M Vekemans, E E E Havinga et al. Sci. Eng., 2001, 32: 1.
[31] L Groenendaal, F Jonas, D Freitag et al. Adv. Mater., 2000, 12: 481~494.
[32] S Akoudad, J Roncali. Synth. Met., 1999, 101: 149.
[33] D J Irvin, C J DuBois, J R Reynolds, Chem. Commun., 1999, 2121.
[34] B De Boer, U Stalmach, C Melzer et al . Synth. Met., 2001, 121:1541~1542.
[35] A Cravino, G Zerza, H Neugebauer et al. Synth. Met., 2001, 121: 1555~1556.
[36] A M Ramos, M T Rispens, J C Hummelen et al. Synth. Met., 2001, 119: 171~172.
[37] S Luzzati, M Scharber, M Catellani et al. Synth. Met., 2003, 139(3): 731~733.
[38] B De Boer, U Stalmach, C Melzer et al. Synth Met., 2001, 121: 1541~1542.
[39] V D V Marleen H, D B Bert, U Stalmach et al. Macromolecules, 2004, 37, 3673~3684.
[40] S M Lindner, M Thelakkat. Macromolecules, 2004, 37: 8832~8835.
[41] X L Chen ,S A Jenekhe. Macromolecules,1996, 29: 6189~6192.
[42] H Krueger, S Janietz, M Thesen et al. Proc. of SPIE, 2004, 5520: 98~109.
[43] S S Sun. Solar Energy Materials and Solar Cells, 2003, 79(2):257~264.
[44] S S Sun, Z Fan, Y Q Wang et al. Proc. SPIE, 2002, 4465: 121~128.
[45] K Tada, M Onoda, A A Zakhidov et al. Jpn. J. Appl. Phys., 1997,36: L306~L309.
[46] S A Jenekhe, S Yi. Appl. Phys. Lett., 2000 , 77(17) : 2635~2637.
[47] M M Alam, S A Jenekhe. Chem. Mater, 2004, 16(23): 4647~4656.
[48] M Granström, K Petritsch, A C Arias et al. Synth. Met., 1999,102: 957~958.
[49] S G Lu, M J Yang, J Luo et al. Macromol. Chem. Phys., 2005, 206:664~671.
[50] H Neugebauer, M A Loi, C Winder et al. Sol. Energ. Mater. Sol. Cells., 2004, 83:201~209.
[51] S L Lu, M J Yang, J Luo et al. Synth. Met., 2004,140: 199~202.
[52] C Winder, G Matt, J C Hummelen et al. Thin Solid Films, 2002, 403-404: 373~379.
[53] 田仁玉,阳仁强,周清梅 等.华南理工大学学报(自然科学版), 2005, 33(2): 6~9.
[54] H Spanggaard, F C Krebs. Sol. Energ. Mater. Sol. Cells., 2004, 83: 125~146.
[55] M Al-Ibrahim, A Konkina, H K Roth et al. Thin Solid Films, 2005, 474: 201~ 210.
[56] 康博南,王立泽,邱勇. 科学通报, 2004, 49: 1611~1613.
[57] J H Hou, C H Yang, J Qiao et al. Synth. Met., 2005, 150:297~304.
[58] B C Thompson, Y G Kim, J R Reynolds et al. Macromolecules, 2005, 38: 5359~5362.
[59] M M Wienk, J M Kroon, W J H Verhees et al. Angew. Chem., Int. Ed, 2003, 42: 3371~3375.
[60] H Krueger, S Janietz, M Thesen et al. Proc. of SPIE, 2004, 5520: 98~109.
[61] B Sun, E Marx, N. C Greenham. Nano Lett., 2003, 3: 961~963.
[62] P Ravirajan, S A Haque, J R Durrant et al. J Appl Phys., 2004, 95: 1473~1480.
[63] J J Dittmer, K Petritsch, E A Marseglia et al. Synth. Met., 1999, 102: 879~880.
[64] M X Chen, E Perzon, N Robisson et al. Synth. Met., 2004, 146: 233~236.
[65] E Perzon, X J Wang, F L Zhang et al. Synth. Met., 2005, 154: 53~56.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号