以成纤维细胞生长因子为靶标的抑制剂的研究进展
- 2012-08-07
- 专题
哺乳动物成纤维细胞生长因子(fibroblast growth factors,FGFs)家族有18 种成员,它们可以结合相应受体(fibroblast growth factor receptors, FGFRs) 并激活下游信号通路,在胚胎发生、发育、血管发生、血管舒张、神经调节、缺血保护、创伤愈合和肿瘤发生等生理和病理过程中起重要作用[1,2]。现已证实,体内FGFs/FGFRs 的过度表达与包括肿瘤(如纤维瘤、神经胶质瘤、黑色素瘤、前列腺瘤、淋巴瘤、白血病、泌尿系统癌等)、骨骼系统疾病(侏儒、颅缝早闭、软骨发育不全、黑棘皮症)和肾衰竭等[1~6]多种疾病密切相关。以FGFs或FGFRs 为靶标的抑制剂具有治疗与FGF/FGFR 过度表达相关疾病的潜能,其中PI-88、苏拉明、PNU145156E 、反应停等以 FGFs 为靶标的抑制剂已进入临床试验[1,6~7]。本文对国内外近年来以FGFs为靶标的抑制剂的研究进展进行了综述。
1 糖类抑制剂
肝素和硫酸肝素(HS)对多数FGFs 的生物活性是必须的:在肝素/HS 和FGF 的共同作用下,FGFR发生二聚化,继而触发靶细胞对FGF 的一系列反应[1,2,8~10]。肝素和HS都是由两种相同二糖单元(α-D-氨基葡萄糖(GlcN)(1→4) ß-D-葡萄糖醛酸(GlcA) 和α-D-氨基葡糖(GLcN)(1→4)α-L-艾杜糖醛酸(IdoA)[14])聚合而成的氨基葡聚糖(GAGs)。HS结合FGF 诱导FGFR二聚化需要糖链长度的最低值为八糖,长度小于8 并能结合 FGF 的肝素寡糖一般表现出对FGF 信号的抑制效应[2,11]。有活性的糖类化合物一般都是通过结合到FGF 的肝素结合位点而抑制FGF 信号[7,11]。
1.1 抑制FGF的糖类化合物
已报道的糖类抑制剂多含有L-IdoA的硫酸化低聚糖[7,12,13]:二糖结构单元为GlcN(2S,6S)(1→4)IdoA (2S)的六糖能强烈抑制FGF-2和其受体的结合,抑制人动脉平滑肌细胞增殖(IC50为16μg/mL)[13];二糖单元为α-D- N -乙酰氨基葡萄糖-(1→4)-α-L-IdoA 的非洲蜗牛粘多糖在体内能够明显抑制FGF-2 刺激的血管增生,抑制鼠黑色素瘤和肺癌的生长[14];4种合成的戊糖(1~4)中,3个糖醛酸残基全为 L-IdoA 的戊糖1比其它3种戊糖(2~4) 的活性(与FGF-2 的结合力、抑制 FGF-2和肝素或HS结合实验、抑制 FGF-2诱导的人动脉平滑肌细胞的有丝分裂)都要强[12]。不含有L-IdoA的糖及糖衍生物也可以结合FGF 并抑制其活性,如未硫酸化的单糖噻唑衍生物 5和6能够明显抑制 FGF-2 和肝素的结合,显著抑制牛动脉内皮细胞的生长[15]。在和FGF 的结合过程中糖残基种类起到主要作用,但带负电荷的硫酸根也起到促进和增强它们结合的作用,甚至一些糖失去硫酸根后对 FGF 无抑制作用[7,11]。过硫酸化的岩藻依聚糖能够抑制FGF-2诱导的人脐血管内皮细胞增殖和血管生成,而脱硫酸的岩藻依聚糖无此活性[16]。Liu 等[11]合成出5种α-D-半乳糖(1→4) ß-D-葡萄吡喃甲苷或者ß-D-木吡喃糖甲苷的二糖(7~11),其中化合物7~10和FGF的亲和力较强,而R3位无磺酸基的 11 活性远低于其它二糖。
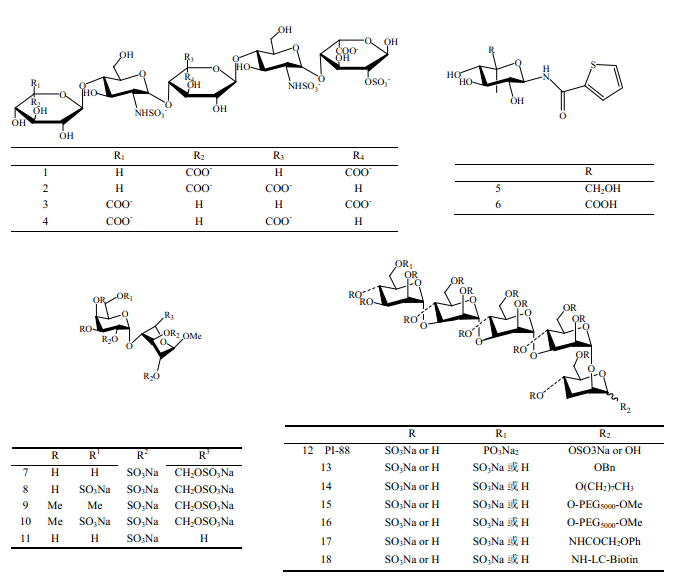
到目前为止,糖类抑制剂方面最有希望的抗肿瘤候选药物是硫酸化磷酸甘露戊糖 PI-88(12),Ⅰ期临床试验表明,它有极好的安全性及耐受性[17~19],在Ⅱ期临床试验中,它辅助治疗肝癌切除显示出良好的抗肿瘤复发作用[17,19]。PI-88的作用机制主要是通过与 HS竞争结合血管生成因子VEGF、FGF-1和FGF-2,阻断生长因子与其受体作用,此外它还可以有效地抑制乙酰肝素酶的活性[17,20]。PI-88对FGF-1和VEGF的结合力比肝素分别高11倍和3 倍,但是对FGF-2 的结合力比肝素低13倍[21]。Karoli 等[24,25]以PI-88为先导化合物,设计合成了19种PI-88衍生物,其中13~18也具有很好的活性,与PI-88相比,它们在拮抗乙酰肝素酶活性方面均保持不变,与VEGF、FGF-1、FGF-2亲和力活性方面也基本保持不变,但在药代动力学方面改变较大,其中1-O-正辛基衍生物(14)的药代动力学有很大提高。
1.2 糖-FGF 相互作用的结构分析
通过对肝素衍生低聚糖-FGF[11]或肝素衍生低聚糖-FGF-FGFR[9,10,23]的X-晶体衍射结构分析发现,肝素衍生低聚糖均是作用于FGF 上带正电荷的肝素作用位点区域,因此,糖上的负电荷基团在相互作用过程中起到重要作用。NMR和X 射线晶体衍射分析发现,肝素和 HS中IdoA残基上的 2-O-磺酸基以及GlcN残基上的N -磺酸基是和FGF 作用的主要基团,GlcN中的6-O-磺酸基仅与FGF-1相互作用,不与FGF-2 相互作用[11,23]。结构单元为 (4-甲基-2-氨基硫酸根合-6-硫酸根合)-α-D-氨基葡糖(1→4)(2-硫酸根合)-α-L-艾杜糖醛酸甲苷的肝素二糖和FGF-1的分子对接数据[11]也证实,糖中磺酸基团和 FGF-1分子中带正电荷的氨基酸残基(Lys112、Lys118、Lys113、Lys128、Arg122、Gln127)相互作用。
2苏拉明类带负电荷的非糖有机小分子
由于肝素上的硫酸根负电荷在结合FGF 的过程中起到了重要作用,因此研究者设计合成了很多带负电荷的非糖有机小分子抑制剂。
2.1苏拉明(Suramin)和Suradistas
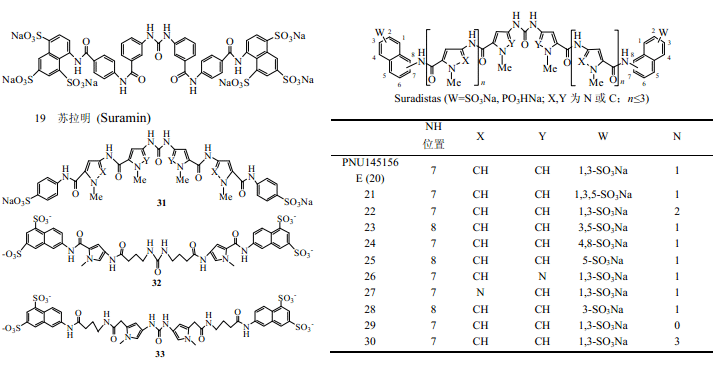
苏拉明(19)为一种含有萘磺酸基的对称分子,是一种抑制 FGF等多种与肝素结合蛋白的生长因子引发的血管增生的药物,可以抑制多种肿瘤细胞的增殖,在临床上用来治疗前列腺癌、膀胱癌等多种肿瘤[7,24~30]。但是由于其在体内的半衰期长(45~55d),有多个可以作用的靶点,可以抑制一系列重要的酶如DNA聚合酶、蛋白激酶C,和血浆蛋白尤其是白蛋白有高亲和力,大剂量使用此药可以引起凝血紊乱、贫血、肾脏病变,从而表现出比较强的毒性而限制了其临床应用[7,24~30]。
Suradistas(20~33)为苏拉明的吡咯和吡唑类衍生物[29],Manetti等合成了75个Suradistas类化合物,并对其进行了抑制FGF-2诱导的Balb/c 3T3细胞的有丝分裂和抑制鸡胚血管增生等活性筛选。高活性的苏拉明及Suradistas分子结构特征及其构效关系可归纳为[7,29~34]:(1)带负电荷的萘磺酸基团可以和FGF中带正电荷的碱性氨基酸相互作用;(2)分子内两端的萘磺酸基团间间距大小对其活性来说很重要,PNU145156E(20,n=1)活性最好,22(n=2)活性也很好,而29(n=0)、30(n=3)的活性急剧降低,原因可能是分子内萘磺酸基团之间的间距只有处于合适长度时,才可以分别与FGF上的肝素结合位点和受体结合位点这两个带正电荷氨基酸的结构域作用;(3)分子的刚性共平面的共轭结构对其活性来说是必须的,整个分子为非共轭结构的化合物(32、33)的活性降低甚至消失,原因可能是共轭结构中断的分子末端萘磺酸基团的摆动自由度增加而导致活性降低。
在Suradistas类化合物中20(PNU145156E)以其高效低毒等优点而进入了II期临床研究[29,30]。它能够高效抑制FGF-2诱导的血管增生、M5076 鼠网状细胞肉瘤、MXT和S180鼠纤维肉瘤等肿瘤的生长,对M5076/CTX和UV2237纤维肉瘤也有抑制作用;它的毒性小于苏拉明,虽可诱导血小板短暂的降低但无肾毒性[32,33,36],具有较好的成药前景。
2.2 其它萘磺酸类化合物
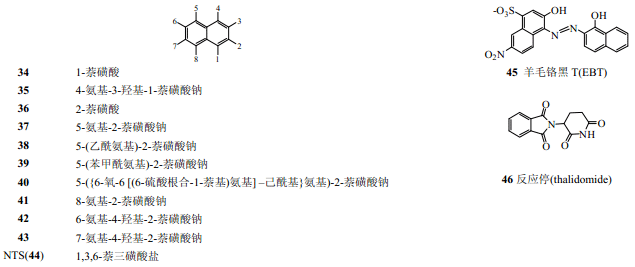
Carlos 等[35]对合成的25个氨基萘磺酸类化合物的活性筛选表明,多个化合物(34~43)都能够明显抑制FGF-1诱导的Balb/c 3T3细胞的有丝分裂活性,其中以35的活性最好;37 的活性仅次于 35,而且它在其抑制有丝分裂的浓度范围内没有表现出细胞毒性[35]。37能够明显抑制小鼠局部缺血视网膜疾病中视网膜新生血管的形成[36],在0.008~0.08mg/kg剂量范围可以抑制皮下埋植含FGF-1或FGF-2的明胶海绵诱导的C57/BI/6鼠新生血管形成,8mg/kg时几乎全部抑制,而苏拉明和 NTS(44)要起到相同抑制作用,一般要求其浓度大于 200mg/kg[35]。像苏拉明一样,37结合FGF有可逆性,肝素可以解除其对细胞增殖的抑制活性[38]。NTS 对FGF-1诱导的血管增生和有丝分裂有高效抑制作用[7,37],可以通过提高细胞凋亡抑制兔角膜新生血管生成和神经胶质瘤生长[38]。羊毛铬黑 T(EBT)(45)在体内也有抑制血管增生和肿瘤生长的作用,在细胞周期的S期阻止人脐静脉内皮细胞增殖,其毒性也低于苏拉明[7,39]。
2.3 抑制剂-FGF 相互作用结构分析
1H-15N HSQC 核磁共振分析显示,苏拉明与hFGF-1的结合位点分布在肝素结合区(Lys127、Tyr139、Lys142)和受体结合区(Leu28、Cys30、Gly34、His35) 两个区域[31]。但分子对接研究显示,肝素结合区和受体结合区的距离大约是3.2nm ,而1个苏拉明分子长度是2.4nm ,因此不可能1个苏拉明分子同时作用在2个位点,而是2个苏拉明分子同时作用在一个蛋白分子表面[31]。用等温滴定量热法确证,苏拉明与hFGF-1按照2׃1 的比例以高亲和力结合[31]。分子排阻色谱分析显示,苏拉明结合到 hFGF-1后形成了1个四聚体[31]。通过稳态荧光检测的热去折叠实验分析显示,苏拉明诱导FGF 聚合分为2步[31],第1步是2个苏拉明分子结合到 1个FGF 上,从而引起了蛋白构象的改变,暴露出了蛋白的疏水表面[31],随后,结合了苏拉明的FGF 通过疏水表面自动形成1个无活性的四聚体,不能再结合 FGFR[31]。
分子对接和分子动力学实验[7,30,33,34]显示PNU145156E 和FGF-1形成1׃1的复合物,通过其萘磺酸基团和FGF-2 上肝素结合位点(Arg121、Lys126、Asn28) 和受体结合位点(Arg108) 两个区域以氢键结合,另外PNU145156E 上氨基和 Pro133、Gly134通过氢键作用。FGF-2 的5 种改构体(Cys87羧甲基化,Lys119Gln、Lys125Gln、Lys129Gln、Lys119Gln–Lys129Gln 定点突变 )对PNU145156E 的离解常数分别提高了12、9.7、9.0、9.1、40.3 倍,而对肝素的离解常数分别提高了0、10、10、10、和100倍,由此可以说明,PNU145156E 可能通过其分子上的萘磺酸基团结合于FGF-2 的肝素结合位点(Lys119、Lys125、Lys129)和Cys87附近的碱性残基(Arg72、Lys77、Arg81、Lys86)[29]。
化合物37(5-氨基-2-萘磺酸钠)和FGF-1 的X 射线衍射晶体数据分析表明[38],37以1׃ 1 的比例直接和FGF-1 分子上的肝素结合位点(Asn32、Lys127、Lys132、Gln141、Lys142)相互作用,前3 个残基通过氢键和磺酸基团作用,后 2 个残基通过疏水作用和萘环作用,另外 37 的氨基通过氢键和Gly140的羰基作用。NOESY核磁共振分析NTS(44)和FGF-1复合物三维结构显示,负电荷的硫酸根和生长因子上正电荷氨基酸Lys127和Lys142相互作用[7,37]。
综上所述,萘磺酸、NTS等小分子均只结合到FGF 上的肝素作用位点,而苏拉明和PNU145156E同时和FGF上的肝素作用位点和受体作用位点发生作用。苏拉明和PNU145156E是结构很相似的 2种分子,但苏拉明-FGF-1和PNU145156E-FGF-2的结合比例分别为2 ׃1和1׃1,这种不同可能有多方面的原因,比如FGF 种类的不同、测定条件不同或者模型都有待进一步完善。总之,糖类拮抗剂-FGF 相互作用的结构数据不是很成熟,还有待于进一步深入研究。
3 活性短肽
有关FGF的肽类拮抗剂报道尚不多,其中大部分都是特异作用于受体(FGFR)膜外部分的短肽拮抗剂[41~47]。Fan等[46]应用噬菌体筛选技术筛选到1个活性15肽(PSPVSELSSGRMAYG),与FGFR1的Ⅱ区180-182和221-223有同源性,能模拟FGFR1上的FGF-1结合区域的三维结构,与FGF-1上的受体结合区域相互作用,抑制FGF-1的促有丝分裂作用。笔者课题组同样采用噬菌体展示技术,筛选到与bFGF 具有高亲和力和最高特异性的小肽P7(PLLQATL),其与FGFR2 D3 256~262这段基序具有较高的序列同源性和相似的高疏水性轮廓,可抑制bFGF 诱导的Balb/c 3T3细胞增殖和鸡胚CAM血管生成[47]。
4 其它
反应停(thalidomide) (46)是一种II 期临床用于治疗FGF-2 诱导的血管增生抑制剂[6],II 期临床用于治疗前列腺癌[48~51]、肾癌[52,53]、黑色素瘤[54]、小细胞肺癌[55]。能够显著降低多发性骨髓瘤病人血浆FGF-2和VEGF的水平,从而降低病人新生血管的生成,是一种很有希望的治疗多发性骨髓瘤的药物[56~59]。研究发现,反应停能够抑制不依赖于贴壁的U-87 MG和神经胶质瘤细胞的生长,而对已经敲除FGF-2基因的相应的贴壁生长的细胞没有抑制作用[60]。因此,其治疗肿瘤的机制为:反应停能够和FGF-2的启动子的富G 区GC boxes (GGGCGG)[60,61]和内核糖体进入位点相互作用[61],因而下调降低 FGF-2的转录和
翻译,降低细胞的FGF-2,从而抑制肿瘤细胞的生长和肿瘤新生血管的形成[61]。
5 结语
FGF 的有机小分子和寡糖类抑制剂多数是通过电荷和FGF 相互作用,因此在体内的靶向性相对较差,毒副作用较大,限制了其应用[1,7]。小肽类抑制剂具有用药剂量小、代谢终产物为氨基酸、毒副作用相对较低等多种优点,但小肽类抑制剂在体内易受肽酶水解而不稳定,从而生物利用度差;小肽的柔性分子结构,可具有多种受体分子识别所需的不同构象,从而使它们的选择性降低,导致药效差,也会产生一些副反应。对短肽进行多种化学修饰后的物质为肽衍生物,这类化合物恰能克服普通短肽的以上缺点,同时又可以兼有普通短肽的优点,近年来肽衍生物的研究已经成为肽类药物研究的一个热点,已经开发出的许多肽类药物都属于肽衍生物,比如抗肿瘤药物奥曲肽等。因此,如果把噬菌体展示技术筛选出来的FGF 拮抗活性小肽改造为肽衍生物,将有可能发现高效低毒的FGF 抑制剂类候选药物。由于已报道的FGF 抑制剂基本多是未经过分子对接等药物设计过程就直接凭经验进行合成和药理研究,所以将来在进行FGF 抑制剂类药物设计时,应该充分利用计算机辅助药物设计等现代药物设计手段,在已经发现的有机小分子抑制剂、寡糖类抑制剂和短肽抑制剂的结构基础上综合分析,设计新的先导化合物。
由于已经确证的多种肿瘤、骨骼系统疾病和肾衰竭等都和体内多种FGF 过度表达有关[1~7],因此对FGF 抑制剂的研究将成为治疗这些疾病的一个重要方向。然而,到目前为止,FGF 抑制剂的研究主要还是集中在FGF-1 和FGF-2 上,对其它的FGF 的抑制剂的研究不多;尽管已经有应用于临床治疗研究的FGF 拮抗剂药物,但是还未发现很理想的高效低毒拮抗剂药物。这一切都预示着对FGF 抑制剂类药物的研究有着非常广阔的空间和发展潜力。
参 考 文 献
[1] A Beenken, M Mohammadi. Nature Rev., 2009, 8(3): 235~53.
[2] M Mohammadi, S K Olsen, O A Ibrahimi. Cytokine, 2005, 16: 107~137.
[3] M Presta, P D Era, S Mitola et al. Cytokine, 2005, 16(2): 159~178.
[4] R Grose, C Dickson. Cytokine, 2005, 16(2): 179~186.
[5] Y H Cao, R H Cao, E M Hedlund. J. Mol. Med., 2008, 86(7): 785~789.
[6] M J Cross, L C Welsh. Trends Pharmacol. Sci., 2001, 22(4): 201~207.
[7] F Manetti, F Corelli, M Botta. Curr. Pharm. Design, 2000, 6(18): 1897~1924.
[8] A D DiGabriele, I Lax, D I Chen et al. Nature, 1998, 393(25): 812~817.
[9] J Schlessinger, Al N Plotnikov, O A Ibrahimi et al. Molecular, Cell, 2000, 6(3): 743~750.
[10] L Pellegrini, D F Burke, F von Delft et al. Nature, 2000, 407(26): 1029~1034.
[11] L G Liu, I Bytheway, T Karoli et al. Bioorg. Med. Chem. Lett., 2008, 18(1): 344~349.
[12] J Kovensky, P Duchaussoy, F Bono et al. Bioorg. Med. Chem., 1999, 7(8): 1567~1580.
[13] Ch Tabeur, J M Mallet, F Bono et al. Bioorg. Med. Chem., 1999, 7(9): 2003~2012.
[14] Y S Lee, H O Yang, K H Shin et al. Eur. J. Pharmacol., 2003, 465(1): 191~198.
[15] A O’ Brien, C Lynch, K M O’ Boyle et al. Carbohydr. Res., 2004, 339(14): 2343~2354.
[16] S Soeda, T Kozako, K Iwata et al. Biochim. Biophys. Acta, 2000, 1497(1):127~134.
[17] V Ferro, K Dredge, L Liu et al. Semin. Thromb. Hemost., 2007, 33(5): 557~68.
[18] L M Khachigian, C R Parish. Cardiovasc. Drug. Rev., 2004, 22(1): 1~6.
[19] R Kudchadkar, R Gonzalez, K D Lewis. Expert. Opin. Investig. Drugs., 2008, 17(11): 1769~76.
[20] L Q Chow, D L Gustafson, C L O'Bryant et al. Cancer Chemother. Pharmacol., 2008, 63(1): 65~74.
[21] S Cochran, C Li, J K Fairweather et al. J. Med. Chem., 2003, 46(21): 4601~4608.
[22 T Karoli, L Liu, J K Fairweather et al. J Med Chem., 2005, 48(26): 8229~36.
[23] M Guerrini, T Agulles, A Bisio et al. Biochem. Biophys. Res. Commun., 2002, 292(1): 222~230.
[24] N Dawson, W D Figg, O W Brawley et al. Clin. Cancer Res., 1998, 4(1): 37~44.
[25] L E Schroder, D Lew, R C Flanigan et al. Urol. Oncol., 2001, 6(4): 145~148.
[26] E M Uchio, W M Linehan, W D Figg et al. J. Urol., 2003, 169(1): 357~360.
[27] X B Zhu, S B Tang, Y Luo et al. Zhonghua Yan Ke Za Zhi , 2005, 41(2): 110~113.
[28] Y Zhang, S Song, F Yang et al. J. Pharmacol. Expt. Thero., 2001, 299(2): 426~33.
[29] M Zamai, C Hariharan, D Pines et al. J. Mol. Struct., 2006, 792-793(3): 23~35.
[30] F Manetti, V Cappello, M Botta et al. Bioorg. Med. Chem., 1998, 6(7): 947~958
[31] K M Kathir, T K Kumar, C Yu. Biochem., 2006, 45(3): 899~906.
[32] K M Kathir, K Ibrahim, T K S Kumar. Biophys. J., 2009, 96(3): 600~601.
[33] M Zamai, V R Caiolfa, D Pines et al. Biophys. J., 1998, 75(2): 672~682.
[34] M Zamai, C Hariharan, D Pines et al. Biophys. J., 2002, 82(5): 2652~2664.
[35] C F Tornero, R M Lozano, M R Horcajo et al. J. Biol. Chem., 2003, 278(24): 21774~21781.
[36] C Lange, Ch Ehlken, G Martin et al. Expt. Eye Res., 2007, 85(3): 323~327.
[37] R M Lozano, M ÁJiménez, J Santoro et al. J. Mol. Biol., 1998, 281(5): 899~915.
[38] P Cuevas, D Reimers, D Diaz et al. Neurosci. Lett.,1999, 275(2): 149~151.
[39] I Langer, G Atassi, P Robberecht et al. Eur. J. Pharmacol., 2000, 399(2-3):85~90.
[40] H K Fan. Prog. Biochem. Biophys., 2001, 28(3): 338~341.
[41] H Fan, Y Duan, H Zhou et al. IUBMB Life, 2002, 54(2): 67~72.
[42] S Kiyota, L Franzoni, G Nicastro et al. J. Med. Chem,, 2003, 46(12): 2325~2333
[43] I Cosic, A E Drummond, J R Underwood et a1. Mol. Cell Biochem.,1994,130(1) :1~9 .
[44] J Ray,A Baird,H G Fred. Proc. Natl. Acad. Sci. USA,1997,94(13) :7047~7052 .
[45] A Yayon, D Aviezer, M Safran et al. Proc. Natl. Acad. Sci. USA, 1993, 90(22): 10643~10647.
[46] H K Fan, Y Liu, H Zhou et al. IUBMB Life, 2000, 49(6): 545~548.
[47] X P Wu, Q X Yan, Y D Huang et al. J.Cell. Mol. Med., 2008, DOI: 10.1111/j. 1582~4934. 2008. 00506.x.
[48] R J Amato, J H McClain, H Henary. Urol. Oncol., 2009, 27(1): 8~13.
[49] S Romero, G Stanton, J DeFelice et al. Urol. Oncol., 2007, 25(4): 284~290.
[50] M C Cox, W L Dahut, W D Figg. Urol. Oncol., 2006, 24(3): 246~249.
[51] R Dreicer, E A Klein, P Elson et al. Urol. Oncol., 2005, 23(2): 82~86.
[52] T Eisen, C Boshoff, I Mak et al. Brit. J. Cancer., 2000, 82(4): 812~7
[53] P E Clark, M C Hall, A Miller et al. Urology,2004, 63(6): 1061~1065.
[54] A B Reiriz, M F Richter, S Fernandes et al. Melanoma. Res., 2004 , 14(6): 527~31.
[55] R F Riedel, J Crawford, F Dunphy et al. Lung Cancer, 2006, 54(3): 431~432.
[56] C S Mitsiades, P J Hayden, K C Anderson et al. Best Pract. Res. Clin. Haematol., 2007, 20(4): 797~816.
[57] A Jurczyszyn, T Wolska-Smole ń , A B Skotnicki. Przegl Lek, 2003, 60(8): 542~7.
[58] F Bertolini, W Mingrone, A Alietti et al. Ann. Onc., 2001, 12(7): 987~90.
[59] W Du, Y Hattori, A Hashiguchi et al. Pathol. Int., 2004 ,54(5): 285~94.
[60] S C Mei, R T Wu. Mol. Cancer Ther., 2008, 7(8): 2405~14.
[61] T D Stephens, C J Bunde, B J Fillmore. Biochem. Pharmacol., 2000 , 59(12): 1489~99.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号