主族元素光控配位数:偶氮苯对主族元素取代基性质的影响和它们的应用
- 2012-08-13
- 专题
1、引述
每个主族元素在主族化合物通常都有它自己的特定的配位数、氧化数和粘结状态。异常的化合价可以通过合适的分子设计和合成方法实现,并且像这样有异常化合价的元素具有独特的特征、特殊的结构和性质。化合物的主要基团的反应能力明显依赖于它们的配位数。例如,硅是14号元素,在周期表中位于碳元素的下方,通常它的配位数像sp3杂化的碳为4。硅也可以处于高配位数状态,例如在合适的条件下,硅在合适的配体中配位数为5或6。在高配位数的硅分子中,处于分子中心的硅原子具有比4配位数硅强的亲电性,并且与硅原子相连的基团的亲核性也随之增强(图1)。因此,一些合成方法已经被开发通过利用高配位数的硅的性质,作为他们的中间体或过度状态,正如已被证实的Tamao氧化反应中表现的性质。1)另外其他例子如有机磷分子中的中心原子P显三价,它的配位数也为3,在过渡金属中已广泛应用于配体。当磷显示五价并且四面体型配位或者显示五价且配位数为5,磷的有效性将大大减少。
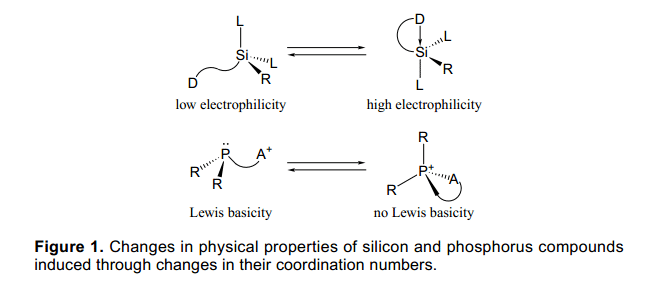
考虑到元素的性质与配位数的关系、氧化态的控制、主族元素的配位数,使用外部刺激很有意义。这将会产生新的方法通过控制物理性质和分子中主要基团性质来控制有机反应。例如,如果配位状态很容易被改变,可以让一个元素在特定阶段表现一种处于激活状态的配位数,而在另一阶段则表现为非激活状态,这将对共聚反应规则非常有用。此外,如果这可以作为原料,它也可以作为一种新的保护方法或脱保护的配体。
然而,主族元素处于非常态的配位状态通常是不稳定的,而在实际应用中有时是需要稳定的状态。例如,空间位阻常被用来来保护低配位数主族元素分子,通过在主族元素上引入体积较大的基团以防止化合物的聚合。此外,处于高配位数状态的元素被认为容易发生消去反应,因为它们在有机反应中常是反应过程中的中间体。因此,需要引入吸电子基团来减弱中心原子的电子密度,并且增强极性键的稳定性,多齿状的配体应该被用于有利的熵。在主族元素分子的非常态配位状态下,分子稳定性的设计中有许多限制因素。为了可逆的控制配位数,控制中心原子与配体之间的关系显得很重要。为了达到和这个目的,作为配体的取代基必须有这些满足这些要求和可控性。因为这些可控性,不可能预先在主要组成化合物上开发出特殊基团来满足接下来的两点要求:1)稳定的异常配位状态;2)结构控制可以由外部因素激活。
一个可控的方法是通过外部因素激活:光照。已有几种分子被证明他们的结构随着光照而改变。在它们当中,偶氮苯作为光敏分子已被广泛报道可以在各种类型的光照反应中被光异性化。2)我们已经将注意力定向在偶氮苯的这个性质上,也就是,偶氮苯的一种结构具备一定的刚度并能感光异构,以及偶氮基团可以起到路易斯碱或利用试剂作为亲电体。我们希望当偶氮苯作为分子内配体时,可以在(E)—和(Z)—之间可逆的光异构化来控制主族元素的配位数。首先,我们在偶氮苯的2号位引入一个主族元素基团,因此,我们可以利用偶氮基团作为光敏点、配位点或亲电点。我们实验的结果是主族元素的碳、硅、磷的配位数在光照条件下可以控制,并且他们的结构、物理性质和反应活性在几种体系中均可很好的光控。在这篇论文中,我们主要关注这几种方法的发展、介绍几种控制主族元素配位数的控制方法以及他们的应用,我们也讨论了偶氮苯自身作为取代基的性质。
2.硅烷和硅酸盐的光控结构以硅元素的配位数为基础
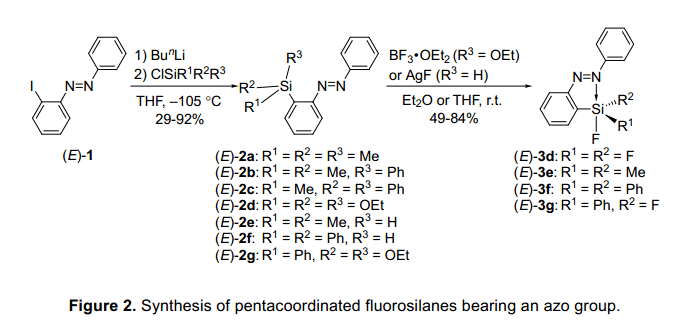
一个2-(苯偶氮基)苯基引入到硅元素上是通过一个有机锂衍生物,它的制备是在-105°C的条件下,2-碘基偶氮苯(E)-1在丁基锂的作用下进行的(如图2)。3,4)硅的配位态是很容易通过29Si的核磁共振波谱确认的。2 -(苯偶氮基)苯基衍生物和与此相似的苯基衍生物的化学位移的差异只有±2ppm,这表明硅元素提供了一个4配位态,其偶氮基团的配位态几乎没有变化。因此,期望硅的亲电性通过氟化作用会增加,单氟硅烷(E)-3e和(E)-3f的制备是通过单氢硅烷(E)-2e 和(E)-2f分别与氟化银反应的。双乙氧基硅烷(E)-2g和三乙氧基硅烷(E)-2d分别与BF3•OEt2反应制取双氟硅烷(E)-3g和三氟硅烷(E)-3d。(E)-3d-g中硅元素的配位态与类似的苯基衍生物通过29Si的核磁共振来相比较,就会发现(E)-3d-g的化学位移高出11 ppm多。科学家发现硅原子的5配位态通过偶碳基团协调作用,氟硅烷衍生物在溶液明显表现出光谱属性。是否存在这种配位作用是经x-线衍射单晶分析确认的 。若化合物中没有氟原子,体积庞大的硅取代基是被发现位于氮原子的对位。通过对比,结果表明,在硅原子上存在至少一个氟原子,五配位态的硅原子可以与一对孤对氮原子相互作用,导致硅原子周围的的周围形成一个扭曲的三方双锥体结构。形成一个N-Si配键诱导代换氟原子是归因于增加亲电性的硅原子和电负性氟原子,正如在前面报道的,有机硅烷的配体数不同于偶氮基团。
硅原子不同的配位状态可以通过化合物的晶体颜色很容易地区别出来。四配位态的含硅偶氮苯且没有氟原子,显示一个红色或橙色的色彩,五配位态的含硅偶氮苯(E)-3d-g显示黄色。通过紫外/可见光谱观察在二氯甲烷的溶液里的四配位态(E)-2a-g化合物,他们的吸收最大值归结为p-p* 和n-p*,分别转换为325-327nm和441-456nm。与没有取代基的偶氮苯(p-p* 318nm ,n-p* 转换为441nm)相比,四配位态(E)-2a-g化合物略红移,这种红移证明通过有机硅基团的供电子基的诱导效应提高了n-和p-轨道的能级。相比之下,在偶氮苯(E)-3d-g与五配位态的硅取代基,最大吸收值归结为p-p*跃迁观察到的红移约336-349nm,而最大吸收值归结为n-p*跃迁观察到相对蓝光偏移。因此,不容易发现化合物(E)-3d或者(E)-3g存在n-p *跃迁吸收光谱,可能是因为这是藏在p-p*跃迁。这些结果表明,氮原子和硅原子形成一个配位键稳定了n轨道并且π轨道变得比π*轨道相对不稳定。
当(E)-3d-g用波长为p-p*跃迁(l=360nm)的紫外灯照射时,使用超高压水银灯和一个彩色的玻璃过滤器,他们异构化成相应的化合物(Z)-3d-g,并且具有81~99%的高收益率(如图3)。异构化作用后,产生这些化合物显示橙色。最大吸收值在大约340nm处下降,与此同时在440nm处有一个最大吸收值,这是因为p-p*跃迁增加了。此外,当(Z)-异构体用波长为l=431nm紫外灯照射,(Z)-异构体能被可逆地转变为(E)-异构体。
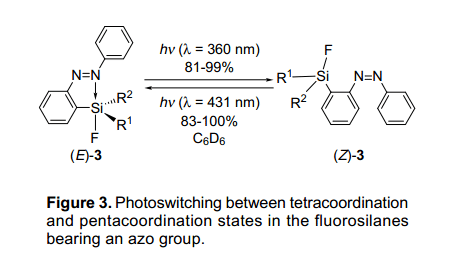
(Z)-异构体在光异构化作用后的配位态可以通过Si NMR检测。(Z)-同分异构体与相应的(E)-同分异构体相比其化学位移要低些,但和苯基衍生物的化学位移相似,这说明(Z)—同分异构体具有四配位态。特别是,氟代甲硅烷基联苯衍生物的两个(E)-3f 和(Z)-3f同分异构体可能被孤立,这表明同分异构体(E)-3f和(Z)-3f的硅原子分别具有五配位态和四配位态。这两种结构通过x-射线衍射单晶分析被进一步证实了,通过光异构化进一步展示了硅原子的结构变化。
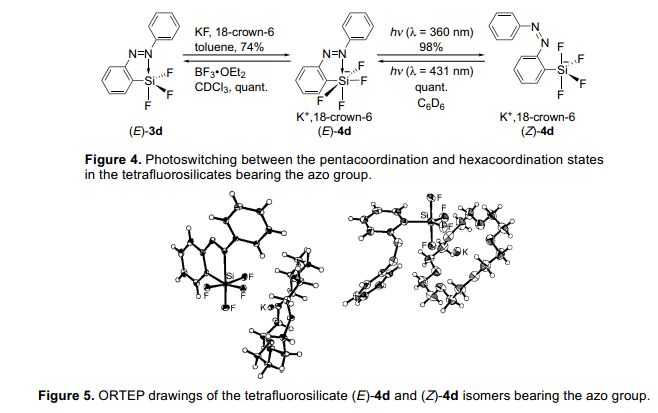
由于我们成功地光转化氟代硅烷的五配位态和四配位态,我们下一步试图通过类似的方法用不同配位态的硅原子开发各种光转化条件。紧接着是五配位态的研究,下一步是研究六配位态。一个硅酸盐和分子配位很容易形成六配位态。我们试图进一步产生更高的配位态的硅原子通过把(E)-3d转换成硅酸盐。我们分析三氟硅烷(E)-3d的氟化作用通过添加氟化钾和18-冠-6醚和已制取的硅酸盐(E)-4d(图4)。5)当用波长类似为偶氮基团的p-p*跃迁的紫外灯照射黄色硅酸盐(E)-4d时,异构化几乎定量进行,形成了红橙色硅酸盐(Z)-4d。异构化的(Z)-4d异构体也定量转变成(E)-4d异构体,暗示(E)-和 (Z)-同分异构体可能定量的进行光异构化。低温条件下19F和29Si核磁共振光谱和x-射线衍射单晶分析表明(E)-4d的六配位态的硅原子有一个扭曲的八面体结构,尽管(Z)-4d五配位态硅原子显示一个三角形的双锥体结构(图5)。最戏剧性的变结构化在被发现在F-Si-F角度,这E)-4d异构体的角度是99.47(5)°和163.73(6)°,虽然(Z)-4d异构体的键角度分别是117.77(19)°和126.67(19)°。此外,(E)-4d,的F-Si-F键角是95.76(5)变为115.53(6)°。这是值得注意的是,光致辐照独自诱导这样一个伟大的结构变化并且在硅原子的周围没有添加任何化学试剂。因此,我们已经成功地通过光致辐照五和六配位态硅酸盐4d控制配位数变化,这一直伴随控制硅原子周围的构象。
有许多的报告中,化合物物理属性的光转化是通过使用偶氮苯的光异构化。在这之前研究,在大多数情况下取代基是位于偶氮基的对位或间位,2)通过调(E)和(Z)同分异构体的每个取代基位置关系或方向,寻找光控条件获得化合物的物理性质。与这些以前光转化系统不同,然而,我们在这里介绍的光转化系统是独特的,硅原子结构可以交换使用在硅和孤对氮原子之间的位置关系的变化。即使化合物的有机轴承取代基的硅原子,如果取代基的方向可以交换通过使用这个方法并且这合成的物质进一步结合定向调控的偶基团的对位取代基,这个系统可能会应用于各种类型物理性质的光转变条件。
3.二氧化硅基于硅原子的配位数变化而导致光转换活性的变化
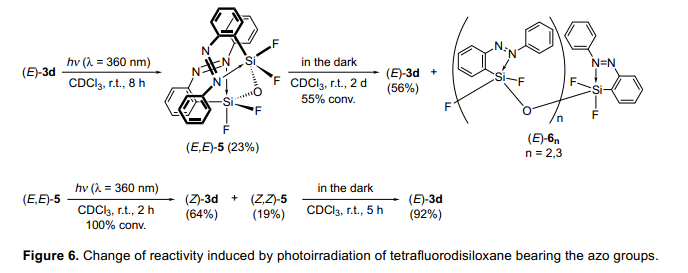
在前面两部分中,我们已经介绍光照可以控制含硅分子的生成结构。那么,接下来的问题已经出现,“我们能否控制他们的反应活性?”当(E)-3d三氟硅烷的稳定性被测定后,研究发现(E)-3d三氟硅烷在(E,E)-5-1,1,3,3,—四氟二甲硅醚渐渐水解了。(图6)(E,E)-5在其分子内连接了两分子的偶氮苯。考虑到偶氮苯可以轻松的堆叠成结晶状态,两分子偶氮苯利用Si-O-Si之间的间隔可以并排在两个苯环的3.4—3.6Å空间内。在避光条件室温下,(E,E)-5二甲硅烷醚在两天内进一步降解,形成(E)3d和(E)-6n(n=2,3)低聚硅氧烷的混合物。光照可以催化(E,E)-5的降解,并且光照2h后,(E,E)-5全部降解,由于这个反应,四配位数的(Z,Z)-5二甲硅氧醚的两个偶氮基团被异构化,除了生成(Z)-3d。当(E,E)-5在避光室温条件下将生成(E)-3d,并且由(Z,Z)-5的合成速率比(E,E)-5的大。
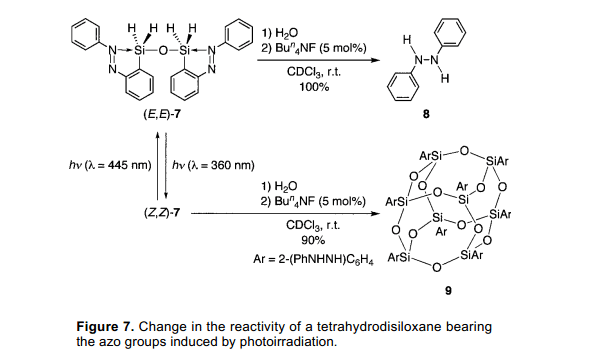
对于(E,E)-7分子,相应的(E,E)-5的四氢衍生物的反应活性在光照前后有明显的变化。在一定量的四丁胺氟化物催化下,(E,E)-7被水解成二苯肼8伴随着C-Si键的断裂。(图7)7)另外,当尝试类似的方法使用四配位数的硅化物(E,E)-7可以定量的光照(Z,Z)-7制得。倍半硅氧烷9的制备没有C-Si键的断裂,尽管偶氮基团减少了。因此,反应活性的改变伴随着C-Si键的断裂可以被光照条件控制。
4.光变对丙烯硅烷反应活性的影响:改变硅原子的配位数
正如在前面章节中展示的,众周所知,有机硅分子的活性因其配位数的不同而明显不同。尽管这里在我们没有详细讨论它们的反应机理,不同的活性伴随不同的配位数有如下原因。相比于电荷形式,硅与取代基键显示极性,三中心四电子键存在于五或六配位数状态。这将导致取代基亲电性的增强,并且取代基活性也比四配位状态强。一般来说,硅原子的配位数可以由四配位状态改良为更高的五或六配位状态,通常是增加一种试剂到反应体系来产生一个高活性的配位物质。然而,正如上面所描述的,控制配位数很容易实现,只需要光照偶氮基团而不需要添加其他任何化学试剂。下一步,我们尝试着控制Hosomi-Sakura反应,这是一个著名的利用硅试剂的反应,并研究光控烯丙硅烷的活性对偶氮基的影响。
具有偶氮基团的烯丙基双氟代硅烷(E)-10被合成,其协配位态通过使用多核磁共振光谱和x-射线衍射单晶分析被研究。正如预期的那样,这是证明(E)-10的硅原子显示五配位态,而光异构化得到的(Z)-10的硅原子显示四配位态。苯甲醛新增了一项亲电基,但是没有反应的进行。因此,在18-冠-6醚存在的条件下,氟化钾与(E)-10反应是为了激活(E)-10,结果导致N-烯丙基硅酸盐11(N-allylated silicate 11)的产生(图8)。另一方面,紧接着(E)-10被光异构化成(Z)-10,其次是在黑暗条件下氟化钾存在于18-crown-6没有反应。有趣的是,光致辐照诱导了反应继续并生成化合物11,类似的例子(E)-10。由于在光致辐照条件下,五配位态的(E)-10和四配位态的(Z)-10可能是相互的转化的;这些结果表明光致辐照条件可以控制烯丙基硅烷的反应性。结果从几个控制的实验显示从(E)-10形成化合物11是一个分子内反应,在这个反应中,烯丙基基团从硅原子转移到氮原子上在g位。这说明,氮配位诱导增加烯丙基基团的亲核性,也增加了硅原子的亲电性,增强偶氮基团的极化并且六元环过渡态都在这些反应的过程扮演着一个重要的角色。
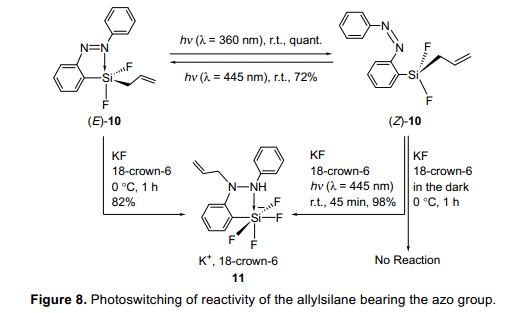
当偶氮集团被认为是一种反应底物,(Z)-和(E)-同分异构体可以分别被认为对应被保护和脱保护的基团,这适用于多步反应包括上面描述的烯丙基化反应。换句话说,保护和脱保护的反应基团是通过使用不同波长的光辐射。
5.硼化合物的刘易斯酸光转化攻击偶氮基团
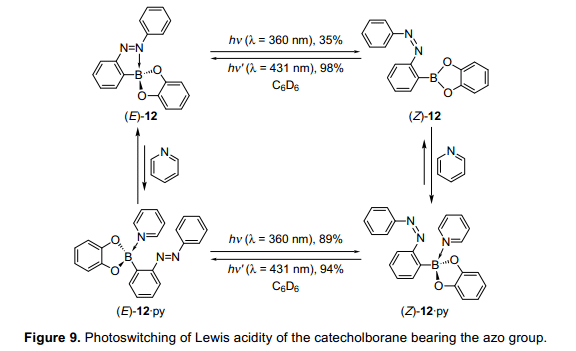
作为主族元素的一个实际应用,除了增加取代基的亲核性,有其他一些方法可以修改主族元素本身的属性,譬如硅化合物。例如,有机硼化合物具有刘易斯酸的性能和该性能已被广泛应用于各种有机合成反应。作为一个基本研究控制与硼化合物反应的反应性例如路易斯酸催化剂,我们调查了光是否控制配位数,儿茶酚硼烷攻击偶氮苯的一部分及其刘易斯酸,评价刘易斯酸的变化使用与吡啶生成络合物作为其反应指数。
硼酸与偶氮苯基团发生脱水缩合反应,生成儿茶酚硼烷(E)-12。光致辐照条件下,(E)-12在苯溶液中可逆地进行光异构化,尽管其异构化效率不高(图9)。等摩尔数的吡啶与儿茶酚硼烷(Z)-12反应,11B核磁共振的结果显示在前场的明显转变,这是证明了一个复杂的成立,吡啶作为一个路易斯碱存在这种异构体。相比之下,当(E)-12的异构体与吡啶反应,化学位移没有显著的变化,表明(E)-12的分子内配位可能会扰乱吡啶分子间配位,导致(E)-12与吡啶不能形成络合物。此外,光异构化反应可逆地进行,甚至在吡啶过量的条件下。即,当用紫外灯照射(Z)-12•py时,(E)-12•py和(E)-12之间的平衡是明显转向(E)-12的。这些结果显示,大多数吡啶分子形成复合物而儿茶酚硼烷可能被释放硼原子和变得自由。当改变等摩尔数吡啶后,通过11B核磁共振估计化学位移值,来测量络合物的络合值。我们发现,络合物增加了大约300倍,通过光异构化使(E)-12转变为(Z)-12。因此,由结果判断与吡啶分子形成络合物,通过刘易斯酸在硼化合物被成功执行。
6.膦基偶氮苯和分子内鏻盐之间的平衡控制
在上面所描述的硅或硼化合物,偶氮基团在这些化合物中担任亲核试剂的作用。然后我们考虑是否有可能控制主族元素的配位数,当偶氮基团也作为一个亲电试剂。有机磷化合物是一个主族化合物作为一个亲核试剂或者一个路易斯碱在一个正常的配位态下。例如,磷化氢已经被广泛的用作与过渡金属形成配体。如果我们能表明我们可以控制有机磷化合物的配位数,那是可以预期,我们也许能够控制化合物的配位能力通过过渡金属和由他们合成的催化剂活性。
2-锂基偶氮苯被二苯氯化磷接受并且膦类化合物的合成尝试通过类似反应使用硅和硼。然而,没有获得相应的膦类化合物。经过反复尝试的过程,我们发现2-碘基偶氮苯(E)-1被转换成磷氢基硫化物(E)-14通过与二苯肼(E)-13反应。(E)-14与三丁基磷化氢经过脱硫作用导致2-双苯基膦基偶氮苯(E)-15的合成(图10)。12)化合物(E)-14也可以得到通过2-锂基偶氮苯与双苯基硫代磷酸化异烟酸氯化物(13)的反应。自膦基基团通常作为一个亲核试剂,但不是一个亲电体,估计偶氮基团可能作为一个电子受体。然而,化合物(E)-15显示一个磷化氢结构,其中在晶体状态中偶氮基团没有交互氮和磷原子。化合物(E)-15在甲苯溶液中显示热致变色和在100°C下显示一个红色的颜色,但在低温条件如-78°C时变成黑色,这样的情况和通常的偶氮苯是不同的。此外,化合物(E)-15的31P核磁共振化学位移表现出明显的温度依赖性。在温度高于60°C时,它在-9.2ppm显示一个单线态,在20°C和-60 °C之间显示一个广泛的信号,并且低于-60°C时在30.1 ppm和-10.3 ppm处呈单谱线。在强磁场信号属于膦结构,而信号低场属于分子内磷盐16基于比较观测到的化学变化和这些估计的GIAO计算。这表明分子内磷盐16应该是分子内磷化氢基团攻击偶氮基团产生的物质。黑颜色是在较低的温度下对应的阴阳离子16的过渡分子内电荷转移的,而红颜色是归因于在高温度下偶氮基团的p-p*跃迁。
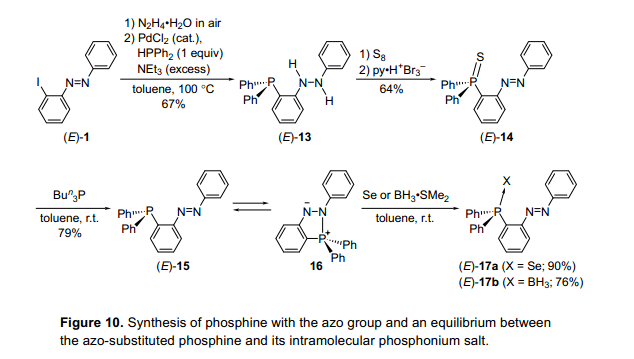
考虑到所有这些结果,在室温下磷化氢化合物(E)-15和分子内磷盐16形成一个平衡。 当温度升高,平衡是转向(E)-15,而低温度导致转向化合物16。这证明了我们可以成功地通过改变温度控制膦(E)-15和磷盐16的平衡。当反应在一个平衡溶液是使用是与硒BH3•SMe2、磷化氢硒化物(E)-17a和磷化氢硼烷(E)-17b反应形成,表明这个磷化氢有普遍的反应性。它可以很容易地与水反应而不像往常膦类化合物。例如,类似的衍生物(E)-15’在4位有一个甲基,存在一个很少量的水促进磷化氢氧化物18的联苯肼基的形成。分子内磷盐来源于2-膦基偶氮苯,显示与Mitsunobu反应的一个中间体相似的性能。14)它们的反应性也支持膦类化合物和分子内季磷盐存在一个平衡。
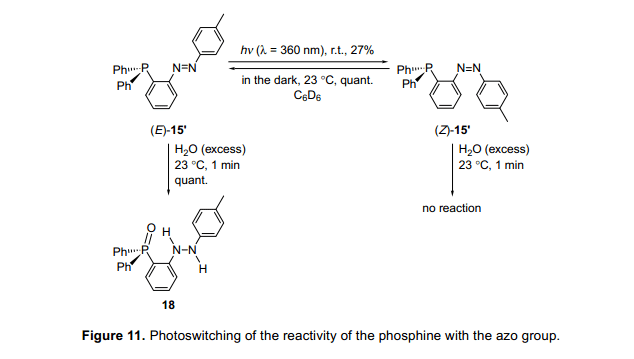
然后,我们怀疑这些膦化合物的配位数可能受光致辐照的控制,光致辐照(E)-15发并现没有异构化发生。然而,有一个甲基(E)-15''经光辐射,光异构化后产生(Z)-15”(图11)。13)然后我们监控(E)-15”或(Z)-15”与水的反应。(E)-15'与水迅速反应,一分钟后完全消失,尽管没有观察到(Z)-15'的变化。最后,在很长的一段时间的反应之后(Z)-15”也产生了18,这是由于它的热异构化。因此,磷化氢的反应性能是在一个有限的时间内可以控制光致辐照分子内磷盐向水均衡。
7.开发的荧光偶氮苯与硼取代基
在前面的章节中讨论的一样,在2位置含有主族元素取代基的偶氮苯显示各种各样的颜色如黄色、橙色、红色和黑色。实际上,通过修改偶氮苯取代基,偶氮苯可以进一步显示各种不同的颜色,并且偶氮苯的这个性能已被用作偶氮染料的基本结构性能。偶氮染料几乎占了全球一半的工业染料生产,并且在印染行业中占据着重要的地位。一些颜料,它被称为荧光染料,不用吸收一个特定光谱的光线,但也能发出光作为荧光。然而,在室温下,偶氮苯一般不会发出任何重要的荧光产生光致辐照,因为光异构化通常在偶氮苯中是收益的。据报道,一些含钯化合物与偶氮苯作为一个配体15)和一些聚合体16)攻击偶氮苯,作为一个组件可以异常发出弱荧光。这含有偶氮苯的钯配合物作为配体,然而,荧光量子收益率只有10-5到10-3。
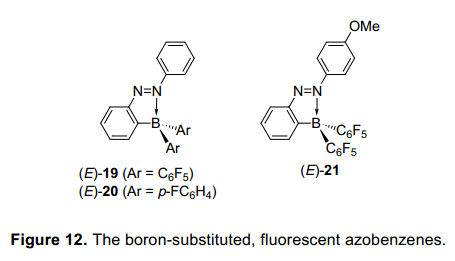
我们已经合成偶氮苯的化合物,这可以接受光异构化,而不受互动的含氮原子的偶氮基团和主族元素的限制。对于含有2—硼取代基的偶氮苯的异构化产量与硅相比很低。这是需要找到一种硼取代基偶氮苯,可以更有效地在光致辐照条件下异构化。在这个随后的研究中,我们调查了大量含偶氮苯基团或硼原子的芳环上不同的取代基,并检查了取代基之间的关系类型和他们的光学性质。因此,我们发现一个平衡在三配位态和四配位态,倾向于转向四配位态,当有一个化合物有供电子偶氮苯基团的4位上或一个吸电子基在硼原子上,这将导致增加可吸收光谱。此外,它也表明了他们配价键增强,异构化率降低,并且有一些化合物未能进行异构化。这些结果表明,硼取代偶氮苯化合物不适合配位数的控制,这也成为我们最重要的目标。然而进一步详细分析了,表明在各种硼取代基的偶氮苯基团不能光异构化,在室内光线下2(五氟苯酚)-硼基-偶氮苯(E)-19在溶液中发出绿色荧光(图12)。17)在室温下在己烷溶液中,荧光量子产率为0.23,这相对偶氮苯是较高的。然而,化合物(E)-20的五氟苯酚基团被对氟苯酚基团所取代,但是不发射任何荧光。这些结果表明,N-B共价键的键能和荧光强度之间可能有关系。因此,我们接下来调查了芳环上的不同取代基,发现化合物(E)-21有一个甲氧基在偶氮苯的4位上具有更强的荧光,并且在室温下己烷溶液中它的量子产率是0.76。在曾被报道偶氮苯衍生物中,这个荧光量子产率值是最高的。它表明,(E)-21发射荧光比未被取代的偶氮苯发射的30000倍还强。
因此,我们下一步试图从理论和实验上确定硼取代偶氮苯化合物发射的荧光原因。这是推测,N-B配位键可能不会只抑制偶氮苯的结构变化,也相对提高p轨道的能级通过降低n轨道的能级。这倒逆转变化能级会使p-p*跃迁从基态跃迁到到最低的能级。因此,从最低的能级跃迁到基态是容易发生的,这进一步导致在一个高产率下吸收光能产生荧光。在最开始的研究中,我们没有设想,引入硼取代基到偶氮苯会诱导荧光发射。然而,这里所介绍的硼取代偶氮苯比报道钯配合物发出更强烈的荧光。主族元素独特的性质提供了偶氮苯的更强烈的荧光,而没有使用昂贵的钯。
8.总结
我们介绍了一个模型系统,可以变换主族化合物的物理性质和结构,通过使用光致辐照诱导偶氮苯的异构化作用,作为一个关键的反应。这种方法同样被认为是适用于其他主族元素,它被高度期望动态地控制元素特定的物理性质而不仅仅是以上描述的那些。如果我们能进一步开发新的方法,可以直接控制主族化合物的配位数,并基于上述阐述的概念,我们相信这种方法的应用将打开一个新方法控制有机反应,这激活/失活方法的使用类似主族元素的配位态的方法。最近报道,一个新的有效合成方法已开发使用,掩蔽和暴露硼元素,18)对于这样的目的如果这里介绍的方法能够得到应用,这个新方法它是可行的。当前的研究结果的理论和实验,并且等同于实际使用和应用在一个在实践层面上。我们认为,然而,我们在这里的概念与传统的是不同的。因此,我们将会很高兴如果这个独特的概念可以用来控制有机反应或化学过程,如保护胺。非常值得一提的是,在研究主族化合物时,我们发现一个意想不到的,令人惊讶的产品——荧光偶氮苯。我们确信主族化合物一定存在很大的研究价值,等待我们去发现和利用。
参考文献
1. (a) Tamao, K.; Ishida, N.; Tanaka, T.; Kumada, M.Organometallics 1983, 2, 1694. (b) Tamao, K., J. Synth.Org. Chem. Japan, 1988, 46, 861.
2. (a) Molecular Switches; Feringa, B. L., Ed.; Wiley-VCH:Weinheim, 2001. (b) Balzani, V.; Credi, A.; Raymo, F.M.; Stoddart, J. F. Angew. Chem., Int. Ed. 2000, 39,3348. (c) Feringa, B. L.; van Delden, R. A.; Koumura,N.; Geertsema, E. M. Chem. Rev. 2000, 100, 1789. (d)Ichimura, K. Chem. Rev. 2000, 100, 1847.
3. Kano, N.; Komatsu, F.; Kawashima, T. Chem. Lett.2001, 338.
4. Kano, N.; Komatsu, F.; Yamamura, M.; Kawashima, T.J. Am. Chem. Soc. 2006, 128, 7097.
5. Kano, N.; Komatsu, F.; Kawashima, T. J. Am. Chem.Soc. 2001, 123, 10778.
6. Kano, N.; Yamamura, M.; Komatsu, F.; Kawashima, T.J. Organomet. Chem. 2003, 686, 192.
7. Yamamura, M.; Kano, N.; Kawashima, T. TetrahedronLett. 2007, 48, 4033.
8. (a) Hosomi, A.; Sakurai, H. Tetrahedron Lett. 1976,1295. (b) Hosomi, A. Acc. Chem. Res. 1988, 21, 200.
9. Kano, N.; Yamamura, M.; Kawashima, T. J. Am. Chem.Soc. 2004, 126, 6250.
10. Yamamura, M.; Kano, N.; Kawashima, T. J. Organomet.Chem. 2007, 692, 313.
11. Kano, N.; Yoshino, J.; Kawashima, T. Org. Lett. 2005,7, 3909.
12. Yamamura, M.; Kano, N.; Kawashima, T. Inorg. Chem.2007, 45, 6497.
13. Yamamura, M.; Kano, N.; Kawashima, T. J. Am. Chem.Soc. 2005, 127, 11954.
14. (a) Mitsunobu, O. Synthesis 1981, 1, 1. (b) Hughes, D.L. Organic Reactions; Wiley: New York, 1992; Vol. 42,pp 335-656.
15. (a) Wakatsuki, Y.; Yamazaki, H.; Grutsch, P. A.;Santhanam, M.; Kutal, C. J. Am. Chem. Soc. 1985,107, 8153. (b) Ghedini, M.; Pucci, D.; Crispini, A.; Aiello,I.; Barigelletti, F.; Gessi, A.; Francescangeli, O. Appl.
Organomet. Chem. 1999, 13, 565.
16. (a) Shimomura, M.; Kunitake, T. J. Am. Chem. Soc.1987, 109, 5175. (b) Han, M.; Hara, M. J. Am. Chem.Soc. 2005, 127, 10951.
17. Yoshino, J.; Kano, N.; Kawashima, T. Chem. Commun.2007, 559.
18. Noguchi, H.; Hojo, K.; Suginome M. J. Am. Chem. Soc.1985, 107, 8153.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号