金催化化学概况及新进展
- 2012-08-22
- 专题
金位于元素周期表的第六周期IB族。正如这个周期的其他后过渡金属,比如铱和铂,它的非同一般的化学催化能力曾经被长期忽视,直到最近才被发掘出来并成为现代化学催化化学发展最快、最具潜力的新兴研究领域,受到有机化学家的广泛关注,在当前有机化学研究热点里唯有有机小分子催化[1]可以与之抗衡。
金的配位化学[2]研究较早,尽管如此,金仍然长期被认为室没有催化活性的金属。早期也有金催化反应的报道[3],但都是被认为和其他金属相比没有任何优越性和不同,这样的状况自1973年开始改观,非均相金催化表现出来的不同一般的催化能力引起了化学家的注意[4];同时,虽然早在1992年Ito[5]等就发表了手行金配体催化的醇醛反应,但是均相金催化直到金催化醇胺对炔烃的亲和加成反应[6]才得以实现。这些分散的报道最终在20世纪末吸引了一些前瞻性强的科学家,他们细致的工作很快打开了金化学的宝藏,由此金化学的“黄金时代”在21世纪初到来,他的秘密迅速被挖掘、解读和拓展。一大批优秀化学家出色的工作不断出现在高端化学杂志上,综述文章也屡见报章[7]。虽然目前金化学研究步伐稍慢,但是突破性的研究还在继续,比如说金钯共催化[8],显示着它旺盛的生命力。
金催化化学大致分为非均相催化和均相催化两类[7a]。本为将专注于对均相金催化做一个描述。迄今为止最详尽的金化学综述当属意大利化学家Areadi在2009年发表与Chem.Rev.的论文[7e];而真正对金化学的反应本质进行深入讨论并提出关键化学理论的综述则是来自于德国化学家Furstner在2007年发表与Angew.Chem.的一篇报道[7b]。在该文中他提出了金离子的“π酸性(π-acidity)”以及相应的“亲碳活性(carbophilic activation)”概念。这些理论,虽然存在一定的模糊性,但是较完美的构建了金化学的初步框架。很明显,随之后来的该领域的进展,很多得益于此。
1 金化学的两个重要概念
1.1π酸性和亲碳活性
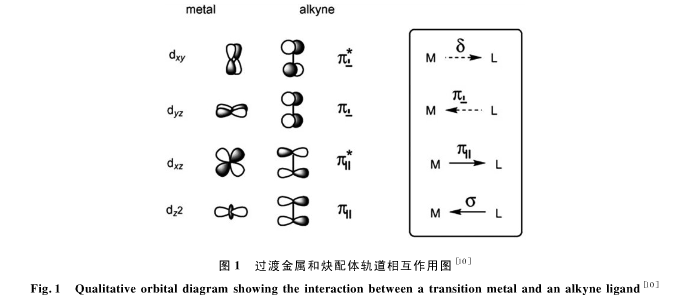
过渡金属和稀炔类π配体形成的络合物的成键形态通常可以用Dewar-Chatt-Duncansan(DCD)模型[9]来诠释。在这个模型里,我、配位键可以看作是电子给体和受体之间的相互关系。在金催化化学领域里,金元素与碳碳叁键配位儿引发的反应更具有起独特性,当然它对碳碳双键的活化[7b]也受到了一定饿重视,特别是对联烯炔烃的活化拓展了联烯化学[7a]。这里,我们通过一价金离子和最简单的叄键配体乙炔的四重电子轨道作用了解释一下金对叄键的活化作用(图1) [10]。精确的计算表明,金离子的d电子轨道和乙炔的π轨道通过轨道对称性形成4层相互作用,其中最重要的L→M(L指配体,M指金属原子)的σ键成键作用的贡献占了65%,而M→L的π键成键作用的贡献则占27%,其他连个作用很弱,只占8%,也就是说,我们可以得出结论:炔烃对于金离子而言是很好的σ给体,同时又是较弱的π受体;这4层作用综合决定了炔烃的电子云密度向金离子转移,也就体现出了金的π-酸性和对炔烃叄键的活化作用。
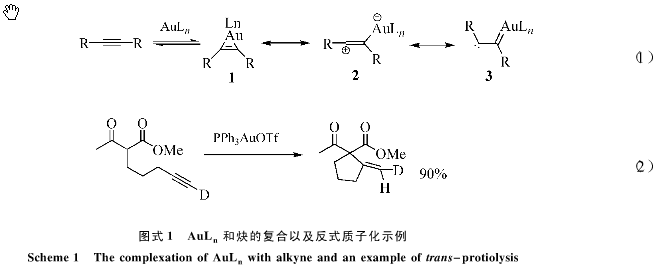
金在与碳碳叄键发生配位作用后可能形成一种金代环丙烯结构1(图式1中的式1),这个结构可以用更接近反应事实的共振结构:金链接的乙烯基碳正离子2来描述;绝大多数金催化的亲核进攻反应在这个阶段,在AuLn从η2向η1配位转化的同时亲核体在反式的方向进攻[11]。Toste等[12]报道的金催化的Conia-ene反应机理研究很好说明了以上理论机制很好说明了以上理论机制(图式1中的式2).在没有诸如醇胺等亲核试剂存在条件下,金离子的电子反馈能力促使电子从金离子中心向乙烯基正离子转移,从而形成金卡宾链接的双自由基中间体3;在众多的烯炔骨架重排反应离可以用这个模型解释金化学的特殊性。
1.2 Fisher-型金卡宾、推-拉反应性和金催化的串级反应
在上面的介绍里面我们已经提到了金卡宾的形成,从头计算表明,金通过起d电子的反馈作用稳定了反应过程中形成的不稳定卡宾[13]。很多实验数据表明,金卡宾不具有碳金双键的特征,儿应当更恰当的描述为金链接的碳正离子。由此,在反应最先引发的时候,金催化剂表现出对叄键“拉”作用,碳碳叄键的电子云密度向金配体转移,而在反应的过程中金的特殊的d电子反馈作用,也就是“推”作用很好的稳定了卡宾。这类缺电子的卡宾极易发生类似频哪醇重排类型的1,2-迁移,从而推动串级重排反应。
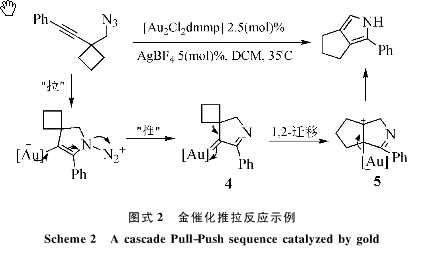
这里可以用一个例子[14]很好的说明以上机制(图式2)。最初的碳碳叄键活化使得起电子被“拉”儿引发叠氮的亲核进攻,儿下一步电子从金配体里面重新释放“推”和氮气单质的形成给出了金卡宾中间体4;环丁烷的环张力通过向金卡宾的1,2-迁移消除环张力形成中间体5。从这么一个典型的例子里面可以看出,完美的设计和对反应机制的预先把握使得反应的流向得到了控制。
在上面的例子中,最后一部金催化剂通过一步电子反馈给正离子从而重新游离出来,实现了催化过程;儿实际上在金催化反应中金配体还可以通过质子化被释放出来。从某种程度上说,金离子和H+可以互相替换,这个过程最常见发生在醇对碳碳叄键的亲核进攻;不同于汞对叄键的活化,碳金键C-Au很容易被切断,从而保证了金催化的高效性[7b];而且它的低毒性和对水和氧气的低敏感性使得金化学受到广泛的欢迎。
以上几个金化学里面经常出现的概念很好的解释了大多数均相金催化的化学反应,巧妙的分子设计和适合反应条件,其中包括最重要的配体的选择,引领出一系列反应结果。当然金化学的本质远不止这些,它还有许多复杂的层面有待化学家去揭示。
2 金化学的两类重要反应
2.1 对碳碳叄键的亲核进攻
亲核进攻反应是一个非常广谱的概念,原则上只要发生了对活化后的缺电子的碳碳叄键的进攻,不管室对一个试剂还是一个官能团,这个过程都被乘坐亲核进攻。因此在金化学领域里面亲核进攻反应占据了很大的篇幅,我们只能做一些简要的描述。其中最具代表的有以下几类:(1)烯炔环异构化反应(Enyen Cycloisomerization) [7e];(2)炔丙酯重排反应;(3)烷氧甲酰化反应(Carboalkoxylation)。
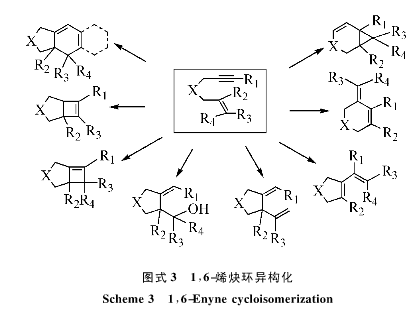
金催化的烯炔环异构化反应发生在一个同时具有碳碳双键和叄键的分子内。它的反应机制曾经困扰了许多化学家,但是随着金催化化学本质的不断揭示,在烯炔结构共同存在下金催化剂有限活化叄键,而烯炔部分则成为亲核试剂;改异构话反应催生了一系列广泛的研究,产生了许多的不同反应途径,极好的体现了金催化化学合成的分子多样性。
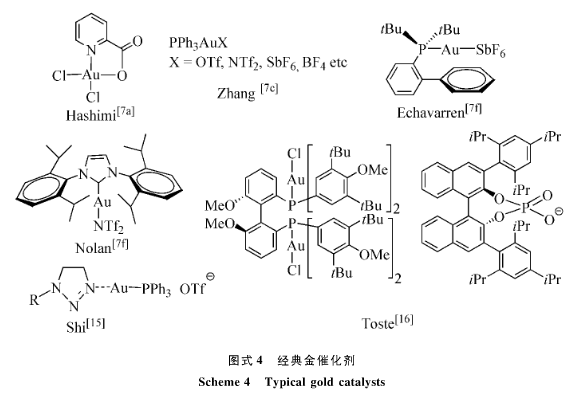
迄今为止有机化学家们已经发展了许多不同配体的金催化剂(图4) [7f],最简单的当属AuCl和AuCl3。2005年Hashimi发现了吡啶-2-甲酸室很好的三价金的配体,相应的络合物(见图4)是个非常温和高效的催化剂,Zhang等成功地拓展了它优异的催化性能;不过使用最广泛的还是稳定的一价金配合物,其中以PPh3AuX和IprAuX为代表,它们都是通过相应的LauCl和银盐的离子交换而增强了金离子的Lewis酸性,其中SbF6-和NTf2-这两个阴离子最为突出。Shi[15]等提出用三唑从动力学上稳定一价金离子的概念;由于一价金离子线性双配位的化学事实,她的不对称催化能力受到很大的限制,为了提高手性选择性,轴手性的双磷配体广泛见于金化学反应;2008年Toste[16]等提出了使用手性阴离子,比如手性磷酸阴离子,与金离子匹配可以有效的增强它的手性环境。尽管金配体化学鸡肋了足够的实验数据,但是目前它的预测能力远不如钯化学,正如烯炔的环异构化反应,它的反应结果总能出人意料!
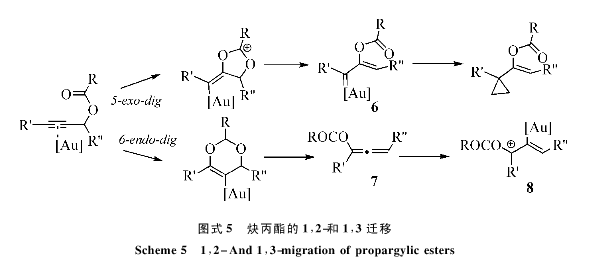
第二类研究很多的炔丙酯的重排反应。简单的炔丙酯回因为末端取代的不同给出不同的反应性:1,2-迁移和1,3-迁移(图5);前者通过金离子的电子反馈能力形成金卡宾6,可以进一步和一个卡宾受体比如烯炔进行环丙化加成反应;后者则通常给出联烯炔中间体7,她可以进一步被进催化剂活化,形成金链接的烯丙基碳正离子8,改中间体可以引发其他串级反应,给出非常有合成价值的分子结构[7a]。
烷氧甲酰化反应通过金催化来实现对叄键的官能团化。该类反应不仅仅包括烷氧化,在Furstner的综述[7b]里把氨化和硫化也都归类在这个反应里面,由此成功的应用于杂环化合物的合成。
2.2 碳氢活化
碳氢活化一直是有机化学家研究的热点,将金催化剂应用于改领域无疑拓展了它的研究空间。然而,在金化学领域内真正的碳氢活化的例子很少。由于目前金催化的碳氢活化反应机理还很模糊,儿通常反应的结果室类似付克产物,因此,在Li等[7d]的一篇金化学综述里面将碳氢活化和金催化的付克反应放在一起讨论。虽然有一些反应食用强的Bronsted酸不能完成转化,但是很多例子表明Bronsted酸也能催化同样的反应。
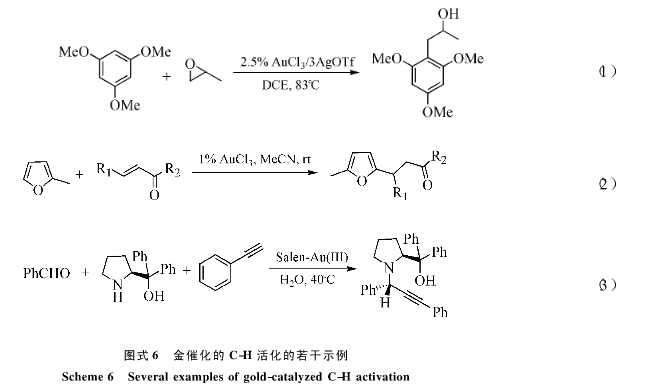
2004年,Shi等[17]报道了AuCl3/AgOTf催化的环氧乙烷的分子内和分子间的氢芳化反应,该反应很可能室通过芳基金对环氧SN2亲核取代的反应机制进行的,给出了和付克反应完全不同选择性的产物(图6中式1);Hashimi等[18]报道的不饱和酮对呋喃的加成产物更有可能室通过付克反应进行的。直观的结论是,芳香化合物的电负性决定反应的机制:电荷丰富的芳香烃更倾向于碳氢活化,而电荷密度较低的则倾向于付克反应(图6中式2)。金催化剂也可以活化Csp-H。也可以活化末端炔氢。一个典型的例子是支等[19]报道的Salen-Au(Ⅲ)催化的水相合成丙炔氨化合物的反应,手性辅助基团使反应具有了大雨99:1的费对映选择性(图6中式3).该类活化也成功的拓展了Sonogoshica偶联反应[20]。
3 金化学的重要进展
3.1 金化学的Suzuki类型的反应
Zhang等[21]在2009年发表于Angew.chem.的一篇论文代表了金化学未来发展的一大趋势:让金也能做类似钯催化的偶联反应。他们发现,一价金可以快速的被腐化试剂1-氯甲基-4-氟-1,4-二氮鎓二环[2.2.2]辛烷的二(四氟硼酸)盐氧化成三价金,而后者可以通过金属交换和还原消除给出偶联产物(图7)。实验表明,烷基金或者烯基金中间体并不是向我们想象中的那样不稳定,它的质子化十一哥相对较慢的过程,也就是说他的软硬度介于钯汞和镁铝之间,使得它有足够的时间被氧化还原,成功的将以上机制应用于碳碳双键的官能团化[22]。

3.2 金催化剂和其他试剂的协同催化反应
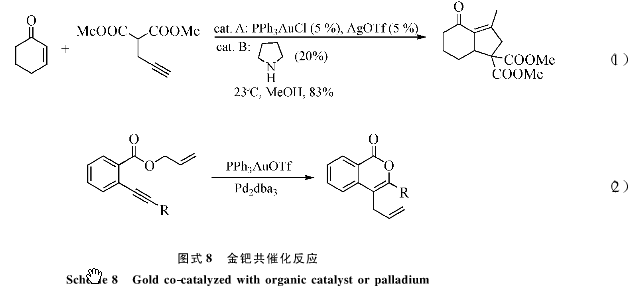
双重催化(Dual catalysis) [23]就是将金对叄键独特活化能力和其他不同类型的催哈ufanying结合起来,比如和有机小分子催化的反应结合起来,从而“一锅法”合成较为复杂的结构分子;而金催化剂其优异的化学选择性和官能团包容性保证了这一策略的实施。例如,一价金催化剂可以很好的和吡咯共同催化(图8中式1)反应[24]。金催化剂还可以和钯试剂共同催化(图8中式2)反应[8]。最新的结果进一步表明,钯可以与金反应中间体进行金属交换,从而让钯接力金完成单纯金催化不能完成的转换。
4.结语
金化学在短短的十几年之内快速发展,已经成为一个十分热门的金属有机化学领域,许多反应被成功发现丙逐步获得应用,同时不断有新的概念被提出和实现,毫无疑问,他的研究和应用价值还将会不断开拓。
参考文献
[1] D W C.MacMillian. Nature, 2008, 455:304~308.
[2] M C Gimeno, A Laguna. Comprehensive Coordination Chemistry Ⅱ,Vol.5,Elsevier, Oxoford. 2004:911~1145.
[3] G C Bond. Gold Bull., 1972,5,11~13.
[4] G C Bond, P A Sermon, D Messy et al. J.Chem. Soc. Chem.Comm.,1973:444~445
[5] Y Ito, M Sawamura, T Hayashi. J. Am.Chem. Soc.,1986,108:6405~6406.
[6] (a)Y Fukuda, K Utimoto.J.Org.Chem.,1991,56:3729~3731;
(b)J H Teles, S Brode, M Chabanas. Angew. Chem.,1998,110:1475~1478.
[7](a)A S K Hashimi,G J Hutehings.Angew.Chem.Int.Ed.,2006,45:7896~7936;(b)A Furstner,P W Davies. Angew. Chem.Int.Ed.,2007,46:3410~3449; (c)L Zhang, J Sun, S A Kozmin. Adv. Synth. Catal.,2006:348,2271~2296;(d)Z G Li,C He, Cbrouwer. Chem. Rev., 2008,108:3239~3265;(e)A Areadi. Chem.Rev.,2008,108:3266~3325;(f)D J Gorin,B D Sherry, F D Toste. Chem. Rev.,2008,108:3351~3378.
[8] Y Shi,K E Roth;S D Ramgren et al. J. Am. Chem. Soc.,2009,131:18022~18023.
[9] M J S Dewar. Rull. Soc. Chim. Fr.,1951,18:C71~C79.
[10] (a)G Frenking, N Frohlich. Chem. Rev.,2000,100:717~774;
(b)A Dedieu. Chem. Rev.,2000,100:543~600.
[11]R Casado, M Contel, M Laguna et al. J Am. Chem.Soc.,2003,125:11925~11935.
[12]J J Kennedy-Smith,S T Staben, F D Toste. J Am. Chem.Soc.,2004,126:4526~4527.
[13] K K Irikura, W A Goddard. J. Am.Chem. Soc.,1994,116:8733~8740.
[14] D J Gorin, N R Davis,F D Toste. J Am. Chem.Soc.,2005,127:11260~11261.
[15] C He, X D Shi, G Novruz et al. J Am. Chem.Soc.,2009,131:12100~12102.
[16] G L Hamitton, E J Kang, F D Toete. Science,2007,317:496~499.
[17] Z J Shi,C He. J Am. Chem.Soc.,2004,126:5964~5965.
[18] A S K Hashimi,L Schwarz, J H Choi. Angew. Chem. Int. Ed.,2000,39:2285~2288.
[19] V K Lo, Y Liu, M K Wong et al. Org. Lett.,2006,8:1529~1532.
[20] Y Fuchita, Y Utsunomiya, M Yasutake. J Chem.Soc. Dalton Trans.,2001:2330~2336.
[21] G Zhang, Y Peng, L Cui, et al.Angew.Chem.Int.Ed., 2009,48:3112~3115.
[22] L Cui, L Cui, Y Wang et al. J Am. Chem.Soc.,2010,132:1474~1475.
[23] A Duschenk, S F Kirsh. Angew. Chem.Int.Ed.,2008,47:5703~5705.
[24] T Yang, A Ferrali, L Camphbellmet al. Chem. Commun., 2008, 61:2923~2929.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号