一种合成含氮化合物的简易方法:Ns-战略和高活性三苯基型树脂
- 2012-11-06
- 专题
1. 介绍
含氮化合物是非常重要的化合物,其中很多都显示出独特的生物活性。鉴于这个原因,在药物活性分子开发领域,它们的有效合成是一个重要的问题。然而,处理这些高极性含氮化合物还有很多问题,此外基础氮化合物氧化性能限制了这些化合物的反应条件。因此,在复杂官能团的合成中为反应中含氮化合物选择保护集团是非常必要的。1)我们发现硝基苯磺酰基团(Ns)烷化反应中为活跃基团,并通过这种方法(Ns-trategy)成功的开发了一种关于二级胺的合成方案。2)此外,温和的条件下可以去除Ns基团。从我们提出来,这种方法已经被很多化学家广泛的的采用,并且我们也利用这个方案完成了具有生物活性的天然产物的全合成过程。3)在利用本方法合成多胺毒素的过程中,我们也成功的开发了一个使用三苯甲基树脂的固相合成步骤。4)使用这种树脂合成相应的固相态代表了一种方法论的进步,因为这其中没有必要的净化过程。本文中介绍关于含氮化合物的一种简单方便的方法。
2.伯胺合成二级胺
胺类的选择性单烷基化是一个不简单的过程,因此二级胺的合成中没有确立单一有效的方法。图表1 显示了一些合成二级胺的典型方法,和每一方法里面存在的问题。例如,伯胺1与烷基卤化物或者磺酸酯烷基化反应不仅仅会得到预期产品二级胺2,由于过烷基化也会生成相应的叔胺3和季铵盐4。同样,醛或酮类在合适的还原剂NaBH3CN等作用下通过还原胺基化生成二级胺5,也会有副产物6生成,当二级胺5中间体产生后,反应有足够的空间位阻和二级等量的羰基化合物凝聚然后还原。用一个强的还原试剂比如LiAlH4和BH3酰化相应的伯胺得到相关的烷基酰胺,然后早通过还原烷基酰胺合成二级胺的方法尽管可行, 但是苛刻的条件阻碍了多官能团化合物合成的应用。近来,关于甲苯砜酰胺8a5) 和三氟乙酰胺8b的Mitsunobu反应已经开发出来。然而, 由于对脱保护(9a,b2)要求条件苛刻,其使用适用范围进限于对碱性介质稳定的化合物。
2.1 2-或者4-硝基苯磺酰胺2)
磺胺类药物是一中可靠的氮保护基,因为他们在强酸或者是常见的碱条件下稳定。此外磺胺类药物的优势在于他们在1 H NMR 图谱中不会发生谱线增宽,而这一现象在相关的氨基甲酸酯和酰胺中是常见的现象。然而它有一个缺点就是去除甲苯磺酰(Ts)和甲磺酰(Ms)这些常用的类似物,要求条件十分苛刻。我们已经证实了再非常温和的条件下相关的硝基苯磺胺类(Ns)可以转换成相应的胺。图表2中,对甲氧基苄胺的反应表明(10)表示一个关于"Ns 战略"的典型的例子,在这个伯胺合成二级胺的反应里面Ns基团既是一个保护基又是一个激活基。

因此,伯胺10和2-硝基苯磺酰氯(NsCI)11a碱参与下反应,得到磺酰胺12a。N-单-取代基磺酰胺12a烷基化反应后很容易与烷基卤化物(R-X)或者是相关的(R-OH)6)在Mitsunobu 条件下反应得产物N-双取代基磺酰胺13a。硝基苯磺酰胺, pKa值很小,烷基化过程中可以作为一个激活基,因此与相对甲苯磺酰化反应相比较为困难的Mitsunobu反应可以顺利进行。当一个温和的亲核试剂(Nu-)比如说PhSH在建德参与下与13a作用, 硫阴离子 亲核加成到苯环上面得到Meizenheimer化合物14a,然后通过去除S02转换成为二级胺。综上所述,Ns 基团温和的条件下可以去除,并且在强酸货碱环境下稳定。因此, 它是一个初级胺或者二级胺的保护基,适用于多个合成反应里。并且, 我们确定外消旋作用在这些反应里面不会发生。
2.2 2,4-二硝基苯磺酰胺(DNs)7)
2,4-二硝基苯磺酰胺(DNs)具有和Ns等同的烷基化能力,但是它有一个缺点就是在建存在下长时间加热会不稳定。然而,它可以选择性的只去除DNs基,因为与需要去除Ns的添加相比他要求的条件比较温和。如图表3所示, 化合物16的DNs基团由两个硝基苯磺酰基团保护着,在HSCH2C02H,Et3N参与下,可以被选择性的去除,然后得到定量的二级胺17。此外,这个过程的一个优点是生成一个副产物2,4-二硝基苯基硫代乙酸酯,the diethyl bicarbonate can be removed is removed by separation of ether and saturated aqueous sodium solution.

3. 受保护的伯胺N-羰基烷氧基硝基苯磺酰胺的合成
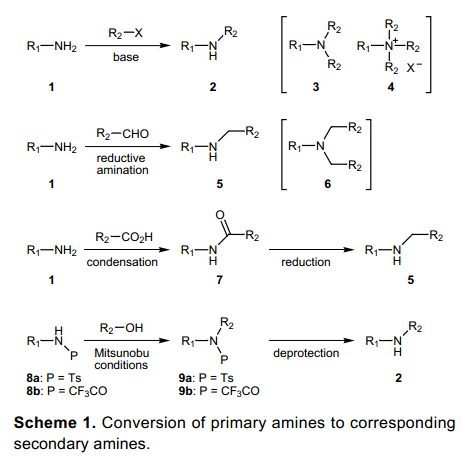
通过取代反应在烷基卤化物或者是醇上面引入一个氮原子来合成伯胺是一个非常实用的反应。尽管Gabriel合成9)和叠氮化物法的方法已经开发出来,仍然有一些常用的直接合成保护伯胺的方法。近来,由Weinreb等人提出的与TsNHBoc相关的Mitsunobu reaction和由Tsunoda10)等人改进的关于TsNH2的Mitsunobu reaction已经被报道。我们认为通过利用Ns 基团的特点可以开发研制更多有用的氮亲核试剂。由NH3和NsCl(11a)很容易制备得到NsNH2,然后在Et3N和一定量的催化剂DMAP参与下,可 以 将 碳酸酯转换得到一个晶体化合物19a的Boc衍生物。这样得到的19a可以顺利的和烷基卤化物烷化反应,和醇进行Mitsunobu反应。这里有一个典型的例子,如图表4所示,19a和(-)-乳酸乙酯单独反应。我们可以从21中选择性的去除Ns基团和Boc基团,因为它本身也是在通常Mitsunobu反应条件下的产物。这就意味着巯基和21反应促使脱去Ns基团, 得到产物N-Boc丙氨酸衍生物22。此外,酸性条件下去除21中的Boc基团可将其促进转换为Ns酰胺23。去除掉22中的Boc基团得到伯胺,通过Ns-strategy可以将23转换为二级胺。此外,用同样的方法可以合成N-Ns-磺酰胺Alloc derivative 19b和Cbz derivative 19c,磺酰胺18和19a-c。
4.分子内烷基化(中型环化合物的合成)
最近,利用烯烃复分解反应合成大型或中型环化合物的研究被大量的报道。然而,也有直接使用氮亲核试剂合成杂环化合物的研究报道。因此,我们采用了Ns基团烷基化完成此类分子间的反应。我们发现他们在八、九、十环合成过程中显示出有用性,这个过程在通常情况下很难完成的。Cs2CO3与卤化物24(磺酰胺18合成所得)反应,加入n-Bu4NI,环化反应顺利进行而后得到杂环化合物25。同样的方法,醇26在Mitsunobu条件下环化得到25 。

5. 天然产物多胺毒素的合成
近来, 一定数量的具有多胺链的天然产物利用微生物技术辅助可以将其分离出来。他们中的大多数常从很少数的天然来源里得到,并且具有很强的生物活性。因此,除了部分二级胺的合成未达满意外,它们已经获得了一个很好的合成收益。我们推断通过和对天然多胺合成的研究,高效的合成方案将会成为可能。
5.1 二胺类的选择性保护
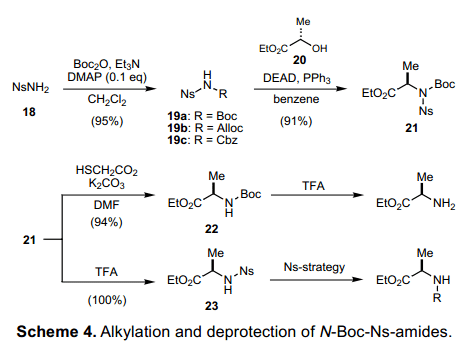
有两个氨基基团的二胺与其他胺之间的区别就是它不仅仅是合成多胺的有用因素,也是可以用作探针分子交联剂的化合物 。然而,对称的二胺类单氨基甲酸酯化进程一般情况下产率很低,且反应产物纯化十分困难。13)我们在其中的一种终端氮中选择性的发现了一种二胺类Ns 基团保护基。如图表6所示,低温下向1,3-二氨基丙烷中缓慢加入NsCl发生单磺酰化反应。单磺酰化二胺27可以通过盐酸与NaOEt中和反应隔离, 滤掉NaCl,蒸发溶剂,减压条件下去除多余的未参加反应的二胺。此外,从含有n=2, 3的二胺中也可能得到高产率的产品28和29。
5.2 HO-416b5) 的全合成
据了解, 从蜘蛛毒腺中提取出来的多聚胺可以抑制刺激性的谷氨酸受体。14)这种受体因为其与大脑神经记忆细胞和思维结构的密切关联,预计可以提供含铅目标化合物用于药物和农药的开发研究。15)我们假设通过Ns-strategy 和从对Holoena curta 分离出来的HO-416b(30)16)的研究中来简单的合成多聚胺。

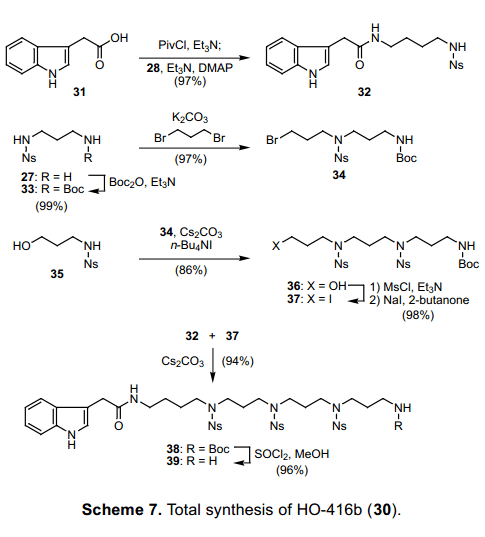
如图表7所示,一个用于合成具有左右独立片段的聚合法被随后设想出来。左片段32通过冷凝吲哚二酸31和二胺28得到。在右片段里面, 通过往Boc引入27的Boc保护基得到二胺33可以用作起始原料。用过量的1,3-二溴丙烷处理可以得到溴化物34, 接着磺酰胺35与其烷基化反应得到36。尽管通过Mitsunobu reaction 和32也可以发生交叉偶联反应,我们选择利用卤化物进行烷基化反应,因为该反应易于净化。醇36mesylation(Ms)后, 得到的中间体与碘37结合,用于和32交叉偶联反应做准备。反应在碱性条件下进展顺利,得到偶联化合物38 ,加入保护基得到伯胺39。
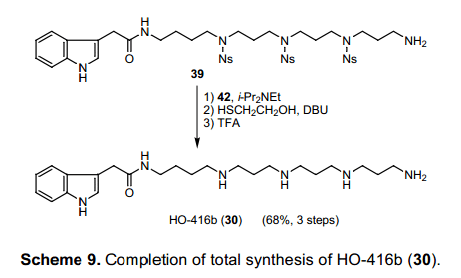
从39中除去Ns基团随后得到HO-416b(30)。然而,不经过辅助物处理去除Ns基团的隔离和净化30的过程是有问题的。一般情况下,反相HPLC和离子交换树脂用于水溶性多胺的纯化。然而,我们认为过剩的试剂和作为副产物的硝基苯衍生物可以通过洗涤祛除,如果Ns 基团的脱保护在固相状态下进行,那么这个净化过程就没有必要。最初我们用相对昂贵的2-氯三苯甲酸树脂40作为伯胺39的一个载体, 然而负载水平不够高不能满足有效的反应。为了提高负荷,我们设计了树脂42,具有远离聚合物的反应点(图表 8). 这个试剂可以通过转换三苯甲基醇41为相应的氯化物制备,用p-羟基三苯甲基醇将廉价的Merrifield resin烷基化而得。树脂42具有很高的反应性能,因为在反应中三苯甲基阳离子受到对位氧原子的保护。此外,这种树脂可回收, 通过复盐化再生并可以重复使用。化合物4,是树脂42的一个稳定的母体。

如图表9所示,39利用i-Pr2Net制备之前负载在固相态。在(HSCH2CH2OH,DBU)条件下完成Ns 基团固相上的去保护过程。洗涤出多余的试剂,干燥树脂后消减(1%,TFA-CH2C12)。祛除溶剂得到纯的HO-416b TFA 盐(30)。通过这个方法,高极性多元胺的隔离就可以不需要通过最后净化固相态去保护作用来完成。
5.3 18元环多胺的全合成, Lipogrammistin-A (43)17)
众所周知大量的含有生物碱的天然产物都具有很大的多元胺环结构。18)尽管很多类似这种目标产物的合成已经有很多的记录,但是具有大环结构的合成还是存在一定的问题。因此 我们推断Ns基团的分子内烷基化反应,不仅仅在合成中等环中是有效的,对合成大环胺类和Lipogrammsitin-A(43)也是十分有用的。

Lipogrammistin-A (43)是一个多聚胺毒素,通过与Tachibana, Fusetani等人一起协作19)从Grammsitidae 表皮结构众分离出来。这个化合物具有一个特殊的结构,包括一个具有18-元环内酯结构的β-氨基酸和一个长的脂肪链。我们全合成过程涉及到分子内Ns基团烷基化的关键步骤如图表10所示。β-氨基酸衍生物45 由已知的光学活性羧酸4420)利用一个Wittig反应作为中心环节通过6步合成得到的。磺胺类45和醇46通过Mitsunobu reaction交叉偶联得到47。因为我们发现烷基磺胺β-消除反应中伴随有甲酯47的基础水解,因此我们选择使用Pd 催化剂保护相应的烯丙基酯,这样也不损害磺胺的完整性。羧酸和二胺衍生物27用混合酸酐法浓缩得到环的先体48。18元环结构,这个过程中的关键一步,是在(Cs2C03, n-Bu4Nl)条件下完成的。
最后, 三个受保护Ns基团与2-甲基丁酸反应完成Lipogrammistin-A(43)的全合成。

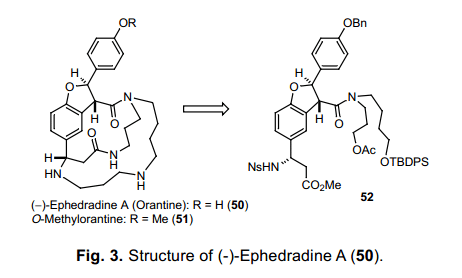
5.4 Ephedradine A (50) 21)的全合成
受到Lipogrammistin-A (43)的全合成良好结果的鼓励,我们的注意力转向Ephedradine A ( 50)的全合成研究方向上,这十一哥更为复杂的结构物。Ephedradine A (50) 是一个精胺生物碱,是由中国传统的药物Ephedra sinica Stapfe 分离出来的有效活性成分。这个化合物从上古年间就已被人知晓,然而它的结构是在1979年由日本东北大学Hikino确定的。22)目标产物由Wasserman于1985年几近合成,不过他们的工作重点集中在O–甲基衍生物的制备上面。这个合成里面最具有挑战性的一点就是在二氢苯并呋喃环不稳定酸和β-氨基酸衍生物不稳定碱参与下合成两个macrolactam 环。另外, 因为Ns-strategy可以促进环化作用并且在温和的条件下去保护基,所以如图3所示,在合成核心部分52光学活性化合物之后用这个方法合成胺类结构成为可能。
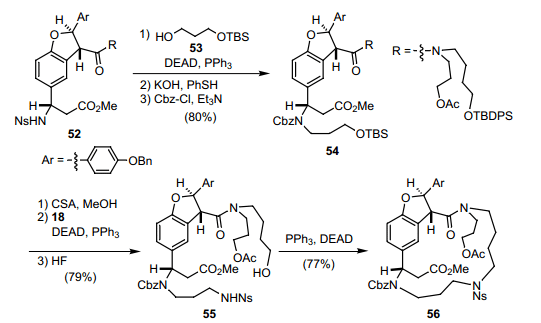
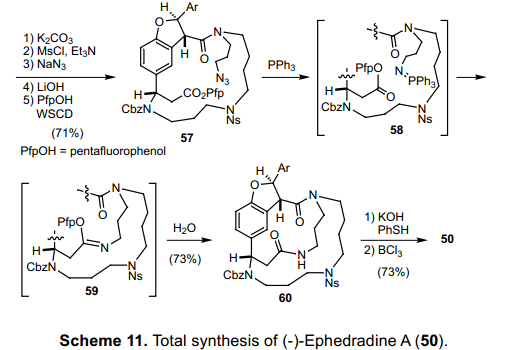
如图表11所示,光学活性二氢苯并呋喃环的进一步中间体52,通过手性C-H插入反应合成得到。24)随后, β-氨基酯通过使用Sharpless手性氨基羟化反应25)作为关键步骤合成得到。After 磺酰胺52加入醇53通过Mitsunobu反应,N原子的保护基团转换伟相对应的Cbz基团,得到产物54。缩合相应的醇和NsNH2(18),接着脱甲硅基反应,移除TBDPS基团,得到成环关键前体55。加入DEAD和PPh3,16元闭环反应平稳进行,得到收益率很好的产物56。随后, 56中的乙酰氧基转换为叠氮化物,然后甲酯转换成五氟苯(PfpOH)酯57。随后57与PPh3发生Staudinger reaction26) 生成不含亚胺基膦苯产物58,然后与活性酯发生分子内aza-Wittig reaction 得到亚胺基乙醚59。这个化合物水解得到 Macrolactam 60。最后,利用BCl3同时除去60中Ns基团中的苄基和Cbz 基来完成 Ephedradine A(50)的全合成。目前这个全合成显示了Ns-strategy 的能量和官能团的范围,这就允许我们来设计演示新的氨基键的化学创造。27)
6. 由高活性三苯甲基树脂(42)完成的固相合成
由我们研发的三苯甲基树脂42,不仅仅具有负荷高效的氨基,而且可以与Ns 氨基和卤代烷烷基化反应,与固相二级胺进行酰基化反应。这个发现为我们研发下述新合成打开了门户。
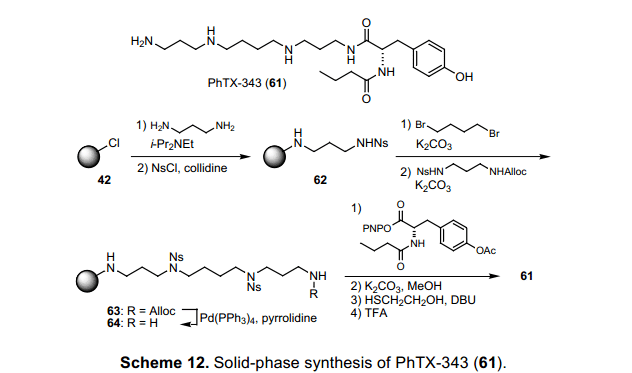
6.1 PhTX-343(61)的固相合成
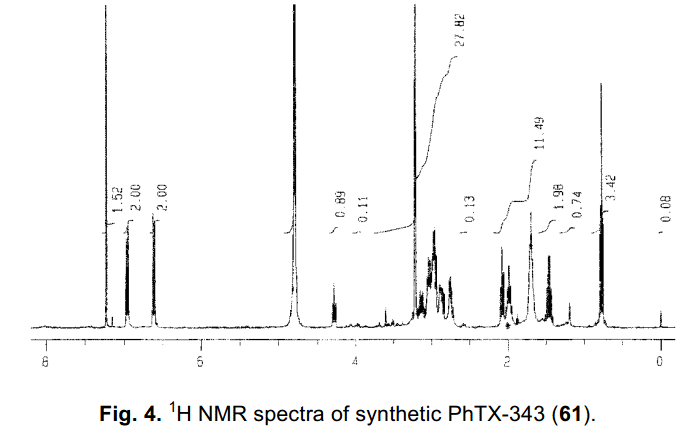
多胺类比如精胺和亚精胺是生命系统中普遍存在的生理活性物质,一定数量的含有这种结构的物质也被大家所熟知.29) 因此,一个合成各种化合物的简易方法备受需要。利用树脂42通过一系列连续反应可以将这个过称完成,并且最终不需要净化提纯。如果固相烷基化得以实现,那么目标产物的综合合成过程就可以成功的实现。为了验证我们这个方法的功效,我们推出了一个精胺衍生物philanthotoxin-343(PhTX-343: 61)的合成实例.30)合成概述如图表12所示。氨基丙烷链接在树脂42之后,游离胺由Ns group保护用于制备Ns-胺62.随后其与二溴丁烷迭代序列通过磺酰胺基的烷基化反应合成精胺衍生物62。根据这个过程,我们可以得出,它有可能与二胺类和卤化物结合,也可以很容易的与醇类发生Mitsunobu reaction。因此,也可以很容易合成一个具有不同长度的聚胺链库。此外,从64中容易移除,得到相应的终极胺可以用于烷基化或者酰基化反应。因此,它是一个用于合成精胺链化合物的很有用的中间体。在PhTX-343(61)的合成中,酪氨酸衍生物引入64中,移除Ns基团,化合物从固相中分离出来完成整个全合成过程,从树脂42开始通过9哥步骤,整个过程收益率为75%。图表4显示材料的1H NMR 图谱,该产品最后一步后去除溶剂而得。在目前的合成方法中,图表4所示利用这个序列,在任何阶段都不需要净化处理,也很有可能获得一个纯的产品。利用这个方法,我们利用固相42和Ns-strategy成功的开发了这个变革性的合成进程。
6.2 平行合成肽型γ–分泌酶抑制剂DAPT衍生物31)
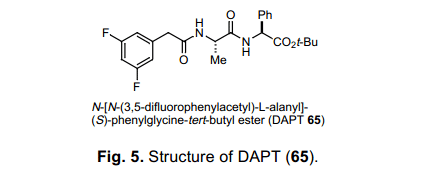
在我们的实验室里面,我们已经研究了γ-分泌酶抑制剂32)对Alzheimer’s 疾病阻缓作用重要性的发展和它的功能性纯化,一个多年来实验室研究合作的项目。33)作为研究的一部分,我们已经完成由Elan Co., Ltd.做出的DATP( 65)34)的结构活性研究,并且发现了活性几乎等量的化合物,其中其C-terminal t-Bu ester 转换伟65中的酰胺基。.
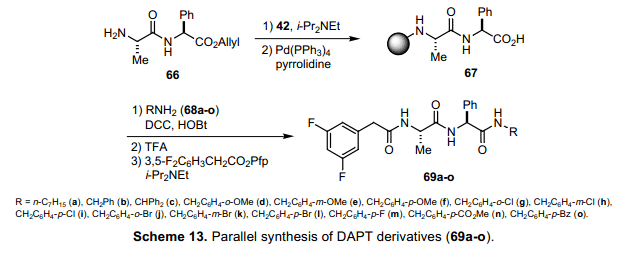
因此,我们试图利用树脂42固相平行合成一系列具有多种C-末端取代基DAPT(65)酰胺化合物。将缩二氨酸链接在固相树脂后,去除烯丙基酯得到羧酸67。在过量的HOBt 存在下使用DOC平缓的将多种胺类68a-o与羧酸67冷凝压缩。酸性条件下从固相中裂解出来,苯乙酸随后引入到活性酯上面。因此,仅仅通过过滤洗涤反应后的粗品,我们成功的得到了具有氨基的衍生物69 a-o,由DAPT(65)C-末端转换而得。
一般情况下,Merrifield method代表性的缩氨酸合成方法通过将C-末端链接到固相上面完成,然后用N-保护氨基酸重复冷凝压缩脱保护。相反,根据当前的合成进程,冷凝压缩过程在利用N-酰基氨基酸末端连接时α-位置不需要差向异构作用。因此,它有可能将肽链由N-末端延长至C-末端。此外,由于事实本身氨基DAPT衍生物69o 比DAPT自身活性强度要大于30倍之多,这项技术在探针的研发过程中是很有效的。
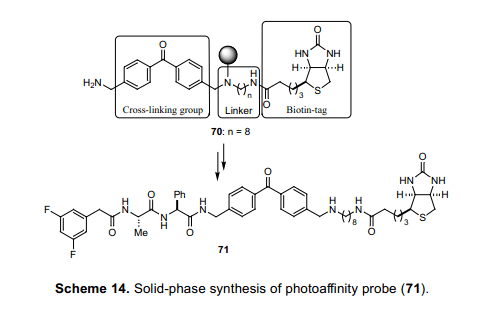
6.3 光亲和探针的固相合成35)
在最近研究的家族病Alzeheimer’s疾病中, 早老素(PS)被克隆是引发这个疾病的原因之一。 后来,具透露, γ-分泌酶本身是一个很庞大的化合物,主要由PS 基因制造的膜蛋白质组成。36)尽管膜蛋白酶生化分析比较困难,但是光亲和标记法是略为有效的方法之一。37)对于探针的合成,引入一个光敏性基团很有必要,这个基团与某种化合物交联,这种化合物见光情况下与高活性蛋白质有很强的亲和力。二苯甲酮是一个常用的光亲和标记用光反应活性基团。因此,这是一个可取的方法,增加了衍生物69o 的活性,显示活性比(65)更高。此外,使用高亲和力生物素的生物素标记法可以更加容易的实现蛋白质的检测与纯化。因此,我们合成和光活性二苯甲酮和蛋白质,同时也合成了固相树脂70,随时可以推出的一种产品。
我们根据上述进程利用单Ns二胺合成物作为连接器。通过Mitsunobu reaction与二苯甲酮链接,生物素浓缩,然后二级胺脱保护Ns基团。这样得到的二级胺链接在固相树脂42上面,以便于成功的合成二苯甲酮-生物素型固相树脂70,该树脂末端处有一个胺基基团可以和配位体反应。随后,浓缩DAPT(65)羧酸衍生物和胺化合物70,我们实现了在酸性条件下通过从固相裂解合成光亲和探针71。这种情况下,就不需要类似合成30 和61案例的复杂的净化过程了。此外,净化此类被引入的生物素非常困难,因为它们不溶于有机溶剂。我们相信,从这个角度来看这个合成方案具有十分重要的意义。事实上,我们已经成功的从早老素中检测出23和26 kDa蛋白质,消失在DAPT(65)参与下利用探针71光亲和标记试验中。38,39)
7. 总结
对于许多药理活性化合物,比如说在生物体内发挥作用的医药产品,溶解度成为一个十分重要和严重的问题,因为它们发挥功能的周围是液态环境。一般情况下,氮原子存在与很多种的医药化合物中间,因为他们在水中显示出很高的溶解性。然而,在目前的报告中,化学家们普遍认为含氮原子化合物的生成多数是有问题的。我们相信使用Ns基团为解决这些问题提供了一个潜在的解决方案,并且已经开发了一个关于利用它的一个广范围的协议。在这个过程里面,我们可以看到Ns group同时有着保护(防御)和烷基化(攻击) 激活的作用。因此,一些天然产物的全合成和某些药物比如说Alzeheimer’s疾病的研发贡献得以实现。总之,我们希望并且期待目前的介绍可以被广泛应用于化学领域。
参考文献
1) T. W. Green, P. G. Wuts, “Protective Groups in Organic Synthesis” 3rd ed., John Wiley and Sons, Inc., New York, 1998, p. 494.
2) (a) T. Fukuyama, C.-K. Jow, M. Cheung, Tetrahedron Lett. , 1995, 36, 6373. (b) W.Kurosawa, T. Kan, T. Fukuyama, Org. Synth. , 2002, 79, 186.
3) Review articles on Ns-strategy, T. Kan, T. Fukuyama, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org Chem. Jpn.), 2001, 59, 779; (b) T. Kan, T. Fukuyama, Chem. Commun., 2004,353.
4) (a) Y. Hidai, T. Kan, T. Fukuyama, Tetrahedron Lett., 1999, 40, 4711. (b) Y. Hidai, T. Kan, T. Fukuyama, Chem. Pharm. Bull., 2000, 48, 1570.
5) (a) J. R. Henry, L. R. Marcint, M. C. McIntosh, P. M. Scola, G. D. Harris, S. M. Weinreb, Tetrahedron Lett., 1989, 30, 5709. (b) T. Tsunoda, J. Otsuka, Y. Yamashita, S. Ito, Chem. Lett., 1994, 539.
6) Review articles on Mitsunobu reaction: (a) O. Mitsunobu, Synthesis, 1981, 1. (b) D. L. Hughes, Org. React., 1992, 42, 335.
7) T. Fukuyama, M. Cheung, C.-K. Jow, Y. Hidai, T. Kan, Tetrahedron Lett., 1997, 38, 5831.
8) T. Fukuyama, M. Cheung, T. Kan, Synlett, 1999, 1301.
9) Review articles on Gabriel method: M. S. Gibson, R. W. Bradshaw, Angew. Chem., Int. Ed., 1968, 7 , 919.
10) (a) T. Tsunoda, H. Yamamoto, K. Goda, S. Ito, Tetrahedron Lett., 1996, 37, 2457. (b) Current review articles on the method of Tsunoda et al.: TCIMAIL, 2004, number123 , 2.
11) (a) T. Kan, H. Kobayashi, T. Fukuyama, Synlett, 2002, 697. (b) T. Kan, A. Fujiwara, H. Kobayashi, T. Fukuyama, Tetrahedron, 2002, 58, 6267.
12) Review articles on natural polyamine: A. Guggisberg, M. Hesse, “The Alkaloids”, Vol. 50, eds. by Cordell G. A., Brossi H. S., Academic Press, New York, 1998, p. 219.
13) W. J. Fiedler, M. Hesse, Helv. Chim. Acta, 76, 1511 (1993).
14) Review articles on spider venom (isolation, decision of structure, synthesis): A. Schafer, H. Benz, W. Fiedler, A.Guggisberg, S. Bienz, M. Hesse, “The Alkaloids”, Vol. 45, ed.by Cordell G. A., Academic Press, New York, 1994, p. 1.
15) Review articles on spider venom (pharmacology): A. L. Mueller, R. Roeloffs, H. Jackson, “The Alkaloids”, Vol. 46, eds. by Cordell G. A., Brossi H. S., Academic Press, New York, 1994, p. 63.
16) G. B. Quistad, C. C. Reuter, W. S. Skinner, P. A. Dennis, S. Suwanrumpha, E. W. Fu, Toxicon., 1991, 29, 329.
17) A. Fujiwara, T. Kan, T. Fukuyama, Synlett, 2000, 1667.
18) Review articles on cyclic polyamines: A. Guggiserberg, M. Hesse, “The Alkaloids”, Vol. 22, 1983, p. 85.
19) (a) H. Onuki, K. Tachibana, N. Fusetani, Tetrahedron Lett. ,1993, 34, 5609. (b) H. Onuki, K. Ito, Y. Kobayashi, N. Matsumori, K. Tachibana, N. Fusetani, J. Org. Chem., 1998, 63, 3925.
20) M. Ohno, S. Kobayashi, T. Iimori, Y.-F. Wang, T. Izawa, J. Am. Chem. Soc., 1981, 103 , 2405.
21) W. Kurosawa, T. Kan, T. Fukuyama, J. Am. Chem. Soc., 2003, 125 , 8112.
22) (a) M. Tamada, K. Endo, H. Hikino, C. Kabuto, Tetrahedron Lett., 1979, 20, 873. (b) P. Dätwyler, H. Bosshardt, S. Johne, M. Hesse, Helv. Chim. Acta, 1979, 62, 2712.
23) H. H. Wasserman, R. K. Brunner, J. D. Buynak, C. G. Carter, T. Oku, R. P. Robinson, J. Am. Chem. Soc., 1985, 107 , 519.
24) W. Kurosawa, T. Kan, T. Fukuyama, Synlett, 2003, 1028.
25) G. Li, H. H. Angert, K. B. Sharpless, Angew. Chem. Int. Ed., 1996, 35, 2813.
26) H. Staudinger, J. Meyer, Helv. Chim. Acta, 1919, 2 , 635.
27) H. Fuwa, Y. Okamura, Y. Morohashi, T. Tomita, T. Iwatsubo, T. Kan, T. Fukuyama, H. Natsugari, Tetrahedron Lett., 2004,45, 2323.
28) T. Kan, H. Kobayashi, T. Fukuyama, Synlett, 2002, 1338.
29) Review articles on polyamines and their coupled body: (a) G.Karigiannis, D. Papaioannou, Eur. J. Org. Chem., 2000, 65, 1841. (b) V. Kuksa, R. Buchan, Synthesis, 2000, 1189.
30) A. T. Eldefrawi, M. E. Eldefrawi, K. Konno, N. A. Mansour, K. Nakanishi, E. Oltz, P. N. Usherwood, Proc. Natl. Acad. Sci. USA, 1988, 85, 4910.
31) T. Kan, Y. Tominari, K. Rikimaru, Y. Morohashi, H. Natsugari, T. Tomita, T. Iwatsubo and T. Fukuyama, Bioorg. Med. Chem. Lett., 2004, 14, 1983.
32) Recent review articles on secretase: M. S. Wolfe, Nature. Rev. Drug. Discov., 2002, 1 , 859.
33) Y. Takahashi, I. Hayashi, Y. Tominari, K. Rikimaru, Y. Morohashi, T. Kan, H. Natsugari, T. Fukuyama, T. Tomita, T. Iwatsubo, J. Bio. Chem., 2003, 278 , 18664.
34) H. F. Dovey, V. John, J. P. Anderson, L. Z. Chen, P. de Saint Andrieu, L. Y. Fang, S. B. Freedman, B. Folmer, E. Goldbach, E. J. Holsztynska, K. L. Hu, K. L. Johnson-Wood, S. L.Kennedy, D. Kholodenko, J. E. Knops, L. H. Latimer, M. Lee, Z. Liao, I. M. Lieberburg, R. N. Motter, L. C. Mutter, J. Nietz, K. P. Quinn, K. L. Sacchi, P. A. Seubert, G. M. Shopp, E. D.Thorsett, J. S. Tung, J. Wu, S. Yang, C. T. Yin, D. B. Schenk, P. C. May, L. D. Altstiel, M. H. Bender, L. N. Boggs, T. C. Britton, J. S. Clemens, D. L. Czilli, D. K. Dieckman-MacGinty, J. J. Droste, K. S. Fuson, B. D. Gitter, P. A. Hyslop, E. M. Johnstone, W. Y. Li, S. P. Little, T. E. Mabry, F. D. Miller, J. E. Audia, J. Neurochem, 2001, 76, 173.
35) T. Kan, Y. Tominari, Y. Morohashi, H. Natsugari, T. Tomita, T. Iwatsubo, T. Fukuyama, Chem. Commun., 2003, 2244.
36) N. Takasugi, T. Tomita, I. Hayashi, M. Niimura, Y. Takahashi, G. Thinakaram, T. Iwastubo, Nature, 2003, 422 , 438.
37) Review articles on photoaffinity labeling: (a) F. Kotozyba-Hilbert, I. Kapfer, M. Goeldner, Angew. Chem., Int. Ed. Engl. ,1995, 34, 1296. (b) G. Dorman, G. D. Prestwich, Biochemistry , 1994, 33, 5661.
38) Y. Morohashi, T. Kan, Y. Tominari, H. Fuwa, Y. Okamura, N. Watanabe, H. Natsugari, T. Fukuyama, T. Iwatsubo, T. Tomita, J. Biol. Chem., 2006, 281 , 14670.
39) H. Fuwa, Y. Takahashi, Y. Konno, N. Watanabe, H. Miyashita, M. Sasaki, H. Natsugari, T. Kan, T. Fukuyama, T. Tomita, T. Iwatsubo, ACS Chemical Biology, 2007, 2 , 408.
注:本文为提供者整理翻译的,由于知识所限,错误在所难免,敬请原谅。如有问题可以查找原文。
快速导航
化学品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
关于物竞
物竞数据库是一个全面、专业、专注,并且免费的中文化学品信息库,为学生、学者、化学品研究机构、检测机构、化学品工作者提供专业的化学品平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、化学品俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们

-
微信账号:物竞化学品数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号